Lowered water table causes species substitution while nitrogen amendment causes species loss in alpine wetland microbial communities
2021-12-22YuntaoLIJinShengHEHaoWANGJizhongZHOUYunfengYANGandHaiyanCHU
Yuntao LIJin-Sheng HEHao WANGJizhong ZHOUYunfeng YANG and Haiyan CHU
1State Key Laboratory of Soil and Sustainable Agriculture,Institute of Soil Science,Chinese Academy of Sciences,71 East Beijing Road,Nanjing 210008(China)
2University of Chinese Academy of Sciences,Beijing 100049(China)
3Department of Ecology,College of Urban and Environmental Sciences,Peking University,Beijing 100871(China)
4State Key Laboratory of Grassland Agro-Ecosystems,College of Pastoral Agriculture Science and Technology,Lanzhou University,Lanzhou 730000(China)
5State Key Joint Laboratory of Environment Simulation and Pollution Control,School of Environment,Tsinghua University,Beijing 100084(China)
6Institute for Environmental Genomics,Department of Microbiology and Plant Biology and School of Civil Engineering and Environmental Sciences,University of Oklahoma,Norman OK 73019(USA)
7Earth Science Division,Lawrence Berkeley National Laboratory,Berkeley CA 94270(USA)
(Received March 1,2020;revised March 20,2020)
ABSTRACT Alpine wetlands are hotspots of carbon(C)storage and methane emission,and they could be key contributors to global warming.In recent years,rapid warming has lowered the water table in alpine wetlands on the Tibetan Plateau, concurrent with intensified nitrogen (N) deposition via anthropogenic activities.We carried out a field experiment to investigate the ecological impacts of these two factors on soil bacterial and functional communities,which are essential drivers of greenhouse gas emissions.Nitrogen amendment alone decreased the phylogenetic alpha-diversity of bacterial communities which could be offset by lowered water table.In contrast,microbial functional alpha-diversity,revealed by a high-throughput microarray,remained unchanged.Both bacterial and functional beta-diversity responded to lowered water table,but only bacterial community responded to N amendment.The alpha-Proteobacteria,beta-Proteobacteria,and Bacteroidetes were the major responsive bacterial lineages,and C degradation,methanogenesis,alkaline shock,and phosphorus oxidation were the major responsive functional processes.Partitioning analysis revealed that N amendment changed bacterial community structure mainly via species loss processes but did not affect bacterial functional communities,with soil pH and ammonium as the key factors influencing changes in bacterial community structure.Conversely,lowered water table altered bacterial and functional communities through species substitution processes linked to soil pH and soil moisture.According to our results,the response mechanisms of microbial communities to lowered water table and N amendment are fundamentally different in alpine wetlands.
Key Words: alpha-diversity,bacterial community,beta-diversity,microbial functional community,N amendment,water table lowering
INTRODUCTION
Among various ecosystems,wetlands are widely considered “the kidneys of the earth”and are fundamental for various biogeochemical cycling processes such as methanogenesis, denitrification, and sulfate reduction (Reddyet al., 1989; Bousquetet al., 2006; Pesteret al., 2012). Although they occupy only 6% of the terrestrial land area,wetlands store approximately 20% of soil organic carbon(C)globally(Mitsch and Gosselink,2007),contributing to approximately 23%of the annual methane production(Conrad,2009).Microbial communities in wetland ecosystems are critical drivers of such processes, playing a primary role in mediating anammox,denitrification,methanogenesis,methane oxidation,and sulfate reduction,in addition to other chemoautotrophy processes(Conrad,1996).Since wetlands are highly sensitive to climate change (Caoet al., 1998),it is critical to elucidate how microbial composition and functional traits respond to environmental changes under future climate change scenarios.
Global warming has altered soil water content and caused the degradation of alpine wetlands(Zhanget al.,2011).Drastic changes in soil water content often affect numerous soil physicochemical properties including soil pH,oxygen(O2)concentration,and redox potential.Notably,bacterial community structure was reported to be niche-oriented across precipitation gradients(Angelet al.,2009),while the alphadiversity remained stable(Angelet al.,2009;Bacharet al.,2010).In sharp contrast,bacterial alpha-diversity fluctuates considerably along a soil water content gradient in agricultural soils(Banerjeeet al.,2016).However,how microbial communities respond to changes in water table levels in wetlands remains unclear.
Nitrogen(N)deposition is increasing due to intensified human activities (Vitouseket al., 1997), which also has considerable impacts on soil microbial communities. Nitrogen fertilization decreases soil bacterial alpha-diversity significantly under both long-term and short-term fertilization in diverse environments ranging from croplands to grasslands (Shen J Pet al., 2010; Zenget al., 2016). A potential mechanism of the decrease is that elevated soil N content stimulates the growth of nitrophilous bacterial communities,which in turn suppresses the growth of other species(Bobbinket al., 2010).Alternatively, increases in ion concentrations in soil could lead to cell destruction and,therefore,decreases in microbial diversity(Treseder,2008).Similarly,N fertilization influences soil functional diversity,as shown in a cucumber and tomato rotation soil study(Shen W Set al.,2010).
There are two major ecological mechanismsviawhich microbial community beta-diversity is altered in response to environmental disturbance,which are referred to as nestedness(also termed as species loss)and species turnover(also termed as species substitution)(Harrisonet al.,1992;Baselga,2010).Species loss occurs when a microbial community is transformed through the extinction of certain species, while the species in the new community can be found in the original one(Ulrich and Gotelli,2010).Consequently, the new community is a subset of the original community. In contrast, species substitution takes place when a microbial community is transformed through the replacement of some of its original species with new species.Therefore,some of the species in the new community may not be found in the original community(Baselga,2010).The two mechanisms have different ecological effects on microbial alpha-and beta-diversities.The species loss changes both microbial community alpha-and beta-diversities.However,if a microbial community undergoes species substitution,beta-diversity increases but alpha-diversity could remain unaltered because the substitution process does not affect species richness.Species loss can result from environmental stress(Waring and Hawkes,2018),selective extinction,or nested habitats (Ulrich, 2009), while species substitution suggests the existence of a geographic or ecological barrier,which selects a certain group of novel species instead of some of the original species (Simpson, 1943). Considering the close correlation between microbial taxonomic diversities and the functional diversity,species loss or substitution would also alter microbial functional community diversity.Species substitution filters certain species that are less competitive when environment changes,and their niches become available for colonization by new species (Aspinet al., 2018). Species that thrive in such a process may be considered specialists since they only adapt to narrow niche ranges(Richmondet al.,2005).Such specialists exhibit species-specific functional traits,including metabolic pathways, substrate utilization, and extracellular product secretion, which influence ecosystem functioning (Dimitriadis and Koutsoubas, 2011). Therefore, the turnover of specialists also results in higher beta-diversity pattern of functional community.However,our understanding of the relationship between functional community diversity and taxonomic community diversity remains poor.To facilitate the understanding of the responses of microbial communities,it is critical to disentangle the distinct effects of the two underlying processes on community-level diversity patterns.Recently,a number of studies elucidating species loss and species substitution processes have focused on animal and plant(Bartonet al.,2013;Heinoet al.,2015;Clarket al.,2017),but rarely on microbial communities.
The aim of the present study was to investigate the impacts of simulated N deposition and lowered water table level treatments on soil microbial communities in a wetland on the Tibetan Plateau (China), the largest and highest alpine ecosystem in the Eurasian continent.Global warming has had a considerable impact on the region, leading to permafrost thawing and wetland degradation (Piaoet al.,2010; Zhanget al., 2011). Low water table levels due to draining activities to meet human water demands also lead to the degradation of wetlands (Chenet al., 2013).In addition, N deposition rates in Tibet have reached up to 17.81 kg ha-1year-1, while the average N deposition rate in 2007 across China was 12.89 kg ha-1year-1(Lü and Tian, 2007). In the present study, we hypothesized that N amendment would decrease bacterial alpha-diversity,which has been observed in numerous previous studies(Campbellet al.,2010;Freedmanet al.,2013;Zenget al.,2016;Dinget al.,2018).We further deduce that microbial functional alpha-diversity would decrease with a decrease in bacterial alpha-diversity. We also hypothesized that N deposition would cause minor or insignificant changes in soil microbial functional community structure.A recent study in soil the Tibetan Plateau demonstrated that soil microbial functional community structure did not change significantly until the N deposition rate exceeded 40 kg ha-1year-1or more(Zhang and Yuan,2015).Considering the simulated N deposition rate in our study may not be high enough to cause significant changes in soil properties,there would be hardly any environmental filtering processes selecting new functional traits.Finally,we hypothesized that water table lowering would influence soil microbial phylogenetic and functional community structure considerably,if the major underlying mechanism is the species substitution process.
MATERIALS AND METHODS
Experimental design and soil sampling
Our study site was located at Haibei Station, to the northeast of Qinghai Province, China (37°29′—37°45′N,101°12′—101°23′E).Soil types and the experimental design have been described previously(Wanget al.,2017).In brief,soil from the Luanhaizi Wetland was collected and then put into bottomless tanks under a rainfall shelter.A randomized block design with two N amendment levels and two water table levels was applied.The two N amendment levels were 0(N0)and 30(N+)kg ammonium nitrate ha-1year-1,and the two water table levels were 3(W0)and-20(W-)cm depth from the soil surface,yielding four treatments:control(no treatment,i.e.,N0+W0),N(N amendment only,i.e.,N++W0),W(water table lowering only,i.e.,N0+W-),and WN(N amendment and water table lowering,i.e.,N++ W-). Five blocks were designed for each treatment as replicates,resulting in 20 plots.The experiment lasted three years from 2009 to 2012. Soil samples were collected on September 12,2012 at a depth of 0—10 cm in each of the 20 plots.After sampling,soils were kept in a cooler and shipped to the laboratory immediately. Soil samples were mixed thoroughly and sieved to remove grass roots and stones,and then divided into two sub-samples. One sub-sample was stored at 4°C for biogeochemical analyses and the other was stored at-40°C for molecular analyses.
Soil physicochemical property analyses
Soil moisture was measured by repeatedly drying 5 g of fresh soil at 105°C until the soil reached a constant weight.The weight ratio of evaporated water to dried soil was determined as SM.Soil pH was measured using a pH meter(Orion Star A111 pH Benchtop Meter,Thermo ScientificTM,Waltham,USA).Soil was suspended in water at a ratio of 1:5(weight:volume)for 30 min and then measured using the pH meter.Fresh soil was air-dried and passed through a 100-mesh sieve for total C(TC)and total N(TN)determination based on dichromate oxidation and titration with ferrous ammonium sulfate.Fresh soil was passed through a 10-mesh sieve for the determination of dissolved organic C(DOC),dissolved inorganic N(DIN),and dissolved total N(DTN).For DOC, the sieved soil was suspended in water (1:10,weight:volume)for 1 h and then sent to a flow analyzer for determination.For DIN and DTN,sieved soil was suspended in 2 mol L-1KCL(1:10,weight:volume)for 1 h and then sent to a flow analyzer for determination. Soil dissolved organic N (DON) was calculated based on the difference between DTN and DIN.
Soil DNA extraction
Total DNA was extracted from 0.5 g soil using a FastDNA®SPIN Kit for soil(MP Biomedicals,Santa Ana,USA)according to the manufacturer’s instructions.Crude DNA was dissolved in 70 μL Tris-EDTA buffer, quantified using a NanoDropTM3300 spectrophotometer(Thermo ScientificTM,Wilmington,USA),and then stored at-20°C.
Bacterial rRNA gene amplicon sequencing
An aliquot(50 ng)of soil DNA was used as a template for PCR amplification.The bacterial V4—V5 regions of 16S rRNA genes were amplified using the following primer set:515F(GTGCCAGCMGCCGCGG)and 907R(CCGTCAATTCMTTTRAGTTT). A unique barcode sequence was added to the forward primer to distinguish samples. Each sample was amplified in triplicate in a 50-μL reaction system under the following conditions: initial denaturation at 94°C for 5 min; 35 cycles of denaturation at 94°C for 45 s,annealing at 55°C for 45 s, and extension at 72°C for 45 s; and final extension at 72°C for 10 min. The PCR products were then purified, mixed together, and sent for high-throughput sequencing using an Illumina Hiseq PE250 platform(Novogene Bioinformatics Technology Company,Beijing,China).
Sequencing data processing
Sequencing data were processed using the Quantitative Insights into Microbial Ecology(QIIME)1.9.0 pipeline(Caporasoet al.,2010b).Sequences were filtered by quality and length control(Phred score threshold 25,length range from 200 bp to 500 bp)and then assigned to each sample.Four samples were discarded due to too few sequences after demultiplexing.After removing barcodes and primers,chimeras were detected and removed using USEARCH(Edgaret al.,2011).Subsequently,sequences were clustered into operational taxonomic units(OTUs)at a 97%similarity threshold using the UCLUST method and representative sequences were selected based on the most abundant sequence in each OTU.To avoid OTU inflation due to sequencing error,only OTUs present in no less than five samples were kept.Taxonomy was assigned to each OTU through their representative sequences using the ribosomal database project classifier(Wanget al.,2007)against the Greengenes database.Representative sequences were aligned using PyNAST,and the aligned sequences were used to construct a phylogenetic tree in FastTree(Caporasoet al.,2010a).Rarefaction was performed to eliminate the impact of different sequencing depths. A randomly selected subset of 32 728 sequences was constructed per sample to compare differences among samples.Microbial alpha-diversity indices(Faith’s PD and observed species)and a beta-diversity index(Bray-Curtis dissimilarity)were calculated in QIIME.
GeoChip analysis and data processing
GeoChip 5.0 was used to determine the functional community structure.Soil DNA was extracted,dyed,purified,hybridized,and scanned according to published procedures(Shenet al.,2016;Wuet al.,2017).The processed GeoChip raw data were filtered further and transformed using the following steps:removing genes detected in less than three out of five samples in each treatment,calculating gene intensity by dividing gene signal intensity by the total signal intensity of the sample, multiplying gene intensity by the average signal intensity across all samples,and obtaining the natural logarithm of each gene intensity.A total of 103 377 genes were examined,and 59 313 genes were retained after quality control.
Statistical analyses
Significant differences in soil properties among different treatments were tested using one-way analysis of variance(ANOVA)in SPSS Statistics 20(IBM Corp.,Armonk,USA).The Bonferroni method was used to conduct multiple comparisons. Multivariate statistical analysis was carried out using the vegan package in R v3.1.3 (R Core Team,2014; Oksanenet al.,2019).Niche width index was used to define how extensively an OTU was distributed within the studied area. Generally, OTUs with low niche width values suggest specialists while OTUs with high values suggest generalists (Fridleyet al., 2007). The spaa package in R was used to calculate the niche width of each OTU(https://CRAN.R-project.org/package=spaa).Two-way ANOVA was applied to test the effects of N amendment and water table lowering on major bacterial phyla and functional groups. A significance level ofP <0.05 was considered significant.Phyla/groups that were tested significant in the two-way ANOVA were further measured for their response value to treatments. To evaluate the response value, we divided the dataset into N+and N0(with or without N amendment)samples across different water table lowering treatments,generating two groups of samples.The response value of each phylum/gene group to N amendment treatment(Res(N),%)was calculated using the following formula:
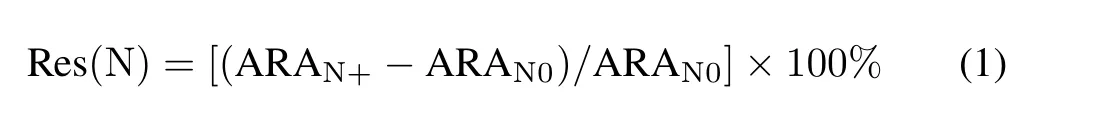
where ARAN+and ARAN0are the average relative abundances of the N+and N0groups,respectively.The response value of each phylum/gene group to water table lowering was calculated similarly.Bray-Curtis dissimilarity based on non-metric multidimensional scaling(NMDS)was used to visualize distances between microbial communities among samples.Permanova analysis(multivariate ANOVA based on dissimilarities) was applied to test the significance of pairwise microbial community differences.Beta-diversity partitioning was performed to measure the extent of species substitution and species loss between microbial communities using the betapart package in R (Baselga, 2010). We used functional substitution and functional loss to describe functional community, in analogy to species substitution and species loss, respectively. Mantel tests were used to determine the correlation between environmental factors and community structure.Figures were made using the“ggplot2”package in R(Wickham,2016)and modified in Photoshop CC(Adobe Systems,Mountain View,USA).
The nucleotide sequence data generated in this study are available in the Sequence Read Archive databases in the National Center for Biotechnology Information(NCBI)website under the accession number SRP136981.
RESULTS
Soil physicochemical properties
Soil pH,SM,and ammonium were significantly influenced by N amendment and water table lowering(Table I)(P <0.05).Soil pH increased from 7.87 in the control to 8.46 and 8.47 in the W and WN treatments, respectively.Soil moisture decreased from 3.59 in the control to 2.54 and 2.50 in the W and WN treatments, respectively. Soil ammonium increased in the WN treatment(110 mg kg-1)compared to the control(53 mg kg-1).No significant changes were observed in other edaphic properties, including TC,TN,C/N ratio,DOC,and DON.
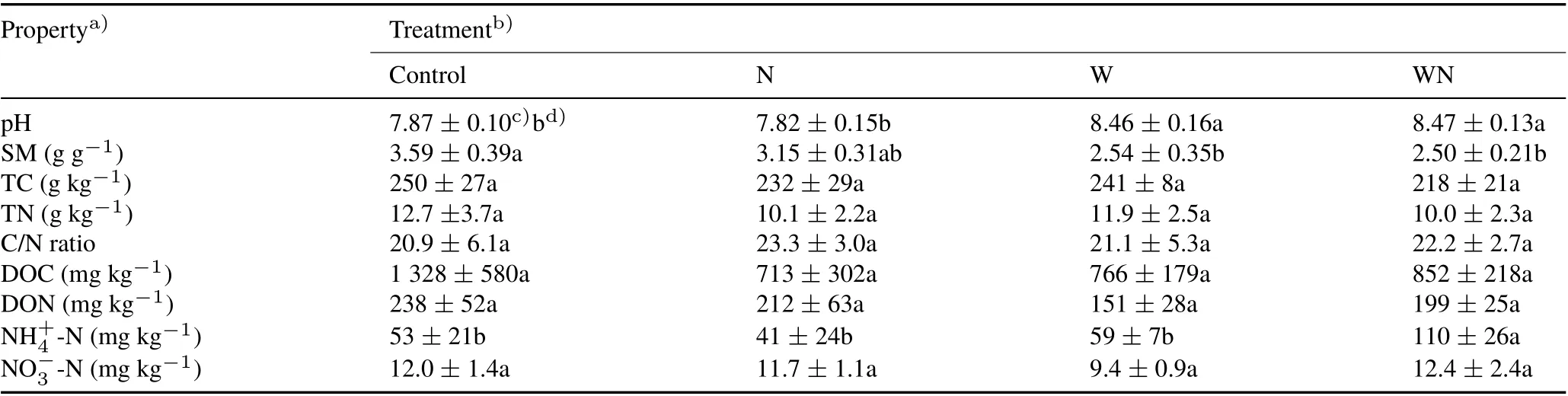
TABLE I Selected soil properties under N amendment and water table lowering
Soil bacterial and functional alpha-diversity
Nitrogen amendment significantly decreased bacterial richness and phylogenetic alpha-diversity (Table SI, see Supplementary Material for Table SI). In contrast, water table lowering treatments(W and WN)did not alter bacterial richness or phylogenetic alpha-diversity. The number of common OTUs were very similar between the treatments and the control(Table SII,see Supplementary Material for Table SII),accounting forca.69%of total OTUs in the control.However,the number of unique OTUs in the N treatment was considerably less than other treatments.Niche width analysis revealed that the N treatment had a similar number of OTUs with high niche width value when compared with the other treatments,but had a lower number of OTUs with low niche width value(Fig.S1,see Supplementary Material for Fig.S1).No significant differences were observed in functional alpha-diversity among the different treatments(Table SIII,see Supplementary Material for Table SIII).
Soil bacterial and functional beta-diversity
Bacterial community composition revealed a clear clustering pattern across different treatments(NMDS analysis,Fig. 1a). In addition, the control and the N treatment revealed a major distinction from the W and WN treatments,implying that water table lowering had a greater impact on bacterial community composition than N amendment.Pairwise comparison revealed significant differences in bacterial community composition for all comparisons except Wvs.WN(Table SIV,see Supplementary Material for Table SIV),with the largest difference between the N and W treatments(coefficient of determinationR2= 0.461) and the least difference between the control and the N treatment(R2=0.230).
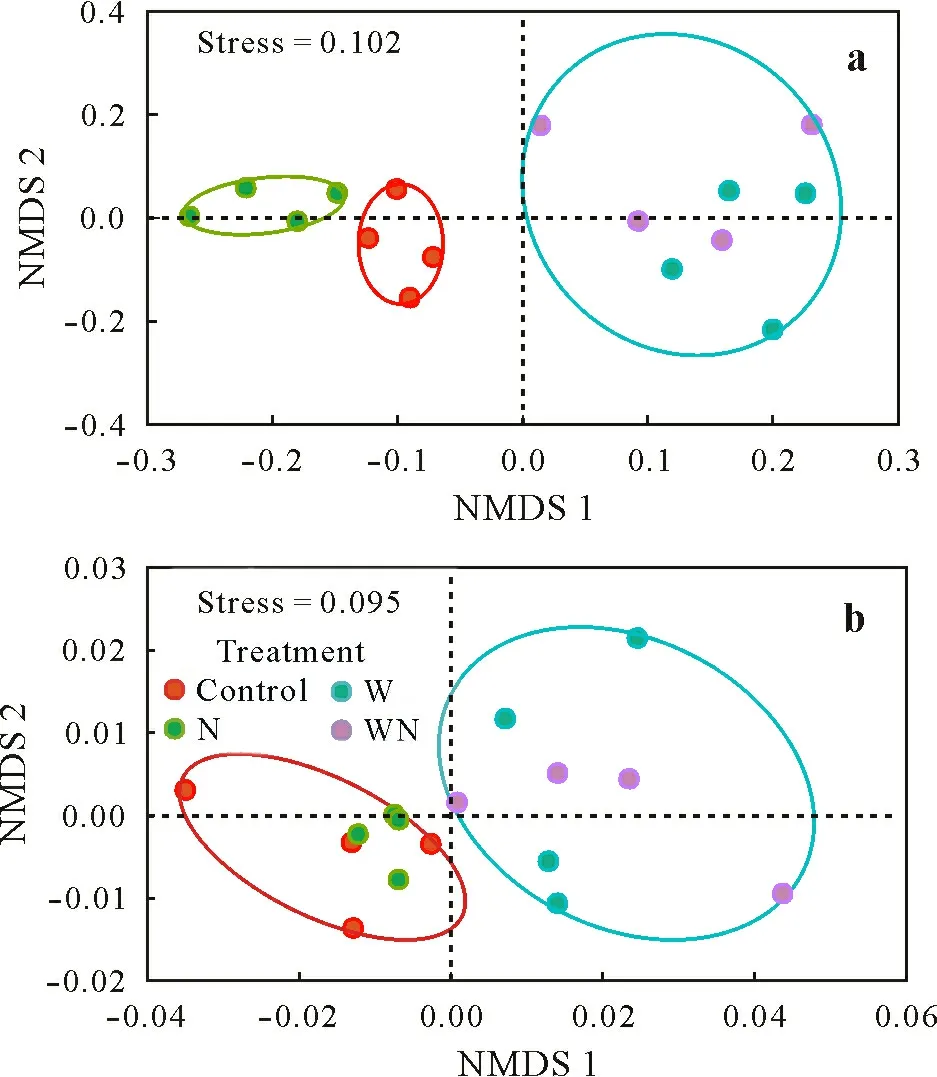
Fig.1 Non-metric multidimensional scaling(NMDS)ordination of soil microbial community structure based on Bray-Curtis dissimilarities of bacterial operational taxonomic units(a)and functional genes(b).Control=no treatment;N=N amendment only;W=water table lowering only;WN=N amendment and water table lowering.
Actinobacteria, beta-Proteobacteria, alpha-Proteobacteria, Bacteroidetes, Chloroflexi, Acidobacteria, delta-Proteobacteria, Firmicutes, gamma-Proteobacteria, and Planctomycetes were the dominant phyla in the bacterial community,accounting for 92.63%of the relative abundance(Table SV,see Supplementary Material for Table SV).Eight phyla responded significantly to N amendment and/or water table lowering(Table SVI,see Supplementary Material for Table SVI).Among them,seven phyla exhibited significant alterations in relative abundance following the water table lowering,while only three phyla were influenced significantly by N amendment (Fig. 2a). No interaction was found between N amendment and water table lowering(Table SVI).Water table lowering significantly decreased the relative abundances of Chloroflexi,beta-Proteobacteria,Chlorobi,and Bacteroidetes by 18.45%(P=0.038),27.96%(P=0.014), 42.79% (P= 0.003), and 55.91% (P= 0.001)respectively, with Bacteroidetes being the most affected phylum(Table SVI).Conversely,water table lowering significantly increased the relative abundances of Acidobacteria,alpha-Proteobacteria,and Planctomycetes by 57.73%(P=0.002), 86.86% (P <0.001), and 108.7% (P <0.009),respectively. In addition, N amendment significantly decreased the relative abundances of gamma-Proteobacteria and Acidobacteria by 19.81% (P= 0.028) and 28.73%(P= 0.012), respectively, while N amendment only increased the relative abundance of Bacteroidetes by 82.06%(P=0.03).The major taxa influenced by treatments were analyzed further(Table SVII,see Supplementary Material for Table SVII).Anaerolineae of Chloroflexi, Bacteroidia of Bacteroidetes,and Ignavibacteria of Chlorobi decreased in the W and WN treatments but not in the N treatment.In addition, the relative abundances of Comamonadaceae of beta-Proteobacteria decreased in the W and WN treatments,but increased in the N treatment.The relative abundances of Hyphomicrobiaceae of alpha-Proteobacteria and Planctomycetia of Planctomycetes increased in the W and WN treatments compared to the control and the N treatment.Acidobacteria-6 of Acidobacteria increased only in the W treatment,while the relative abundance of Xanthomonadales of gamma-Proteobacteria was the highest in the N treatment,with no differences observed among the control and the W and WN treatments.
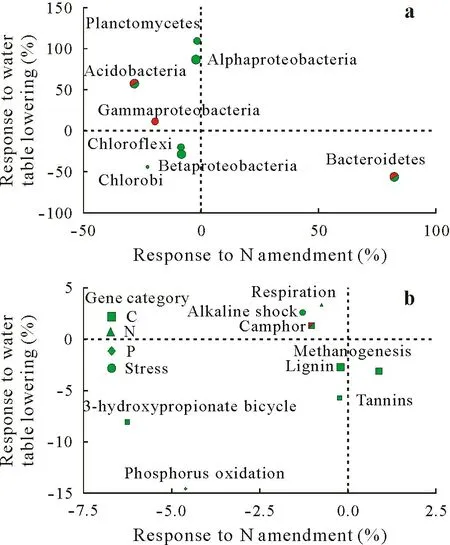
Fig. 2 Responses of soil major bacterial phyla (a) and functional gene groups(b)to N amendment and water table lowering.Responses of each phyla/group were based on variations in relative abundance and are presented as percentage changes.Responses to N amendment and water table lowering are shown orthogonally. Red, green, and bi-colored points indicate a significant response to N amendment, water table lowering, and both treatments,tested by two-way analysis of variance,respectively.Point size denotes the square root of the sum of relative abundance across all samples in each phyla/gene group.
Similarly,functional communities of the control and the N treatment and those of the W and WN treatments were clustered separately(NMDS analysis,Fig.1b).The N and WN treatments showed the largest differences in community structure (R2= 0.352, Table SVIII, see Supplementary Material for Table SVIII), with the smallest differences observed between the control and the W treatment(R2=0.219).Functional community structure in the N treatment did not vary from that in the control.When NMDS analysis was applied to C cycling,N cycling,phosphorus(P)cycling,and stress genes, similar patterns were observed(Fig.S2,see Supplementary Material for Fig.S2).
Metal homeostasis was the most dominant functional gene category in relative abundance,followed by the C cycling,stress,virulence,organic remediation,and N cycling,which collectively accounted for 83%of the relative abundance(Table SV).Genes associated with the biogeochemical cyclings of C, N, P, and sulfur, as well as stress response genes, are summarized in Fig. 2b. The N treatment only increased the relative abundance of camphor genes belonging to C degradation. Eight sub-categories were altered by water table lowering,including decreases in lignin and tannis degradation genes, 3-hydroxypropionate bicycling genes related to C fixation,methanogenesis,and P oxidation genes,and an increase in alkaline shock genes(Table SVII).
Environmental factors correlated with soil bacterial and functional community structure
Bacterial community structure was significantly correlated with soil pH(the strongest correlation,correlation coefficientr=0.655)and SM(r=0.430)(Mantel tests,Table SIX, see Supplementary Material for Table SIX).Similarly,soil pH and SM were correlated significantly with functional community structure on the whole and biogeochemical cycle genes,with soil pH being the predominant factor(Tables SX and SXI,see Supplementary Material for Tables SX and SXI).
The underlying mechanisms shaping soil bacterial and functional community structure
Beta-diversity was partitioned into community species substitution and species loss to discern the different underlying mechanisms shaping bacterial community structure.The N treatment led to significant community species loss(R2= 0.723) compared to the control, while the W and WN treatments did not significantly influence community species loss(frequency distribution of beta-diversity,Fig.3).Community species loss was correlated significantly with soil pH(r=0.411,P=0.003)and soil ammonium(r=0.500,P=0.004)(Mantel tests,Table II).In sharp contrast,species substitution was significantly induced in the W(R2=0.198)and WN(R2=0.197)treatments,but not in the N treatment.In addition,species substitution was significantly correlated with soil pH (r= 0.260,P= 0.007) and SM(r=0.477,P=0.001).
Functional loss did not vary significantly among treatments(Fig.3).The W and WN treatments both significantly influenced community functional substitution,while the N treatment did not.Mantel tests(Table II)detected no significant correlation between soil factors and functional loss.Soil pH,SM,and DON were significantly correlated with functional substitution,with pH having the greatest effect(r=0.241).
DISCUSSION
Soil bacterial and functional alpha-diversity
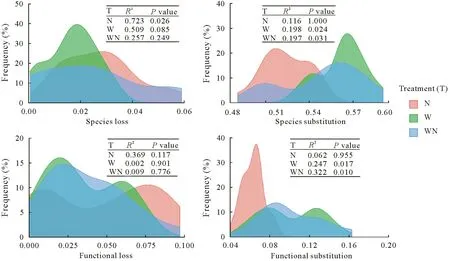
Fig.3 Frequency distribution of partitioned beta-diversity components for soil bacterial and functional communities.Each distribution was generated by plotting the corresponding beta-diversity components between N amendment alone(N),water table lowering alone(W),or in combination(WN)treatment and control(no treatment).Permanova statistic coefficient of determination(R2)and P values are annotated to each distribution to test significance of community variation.
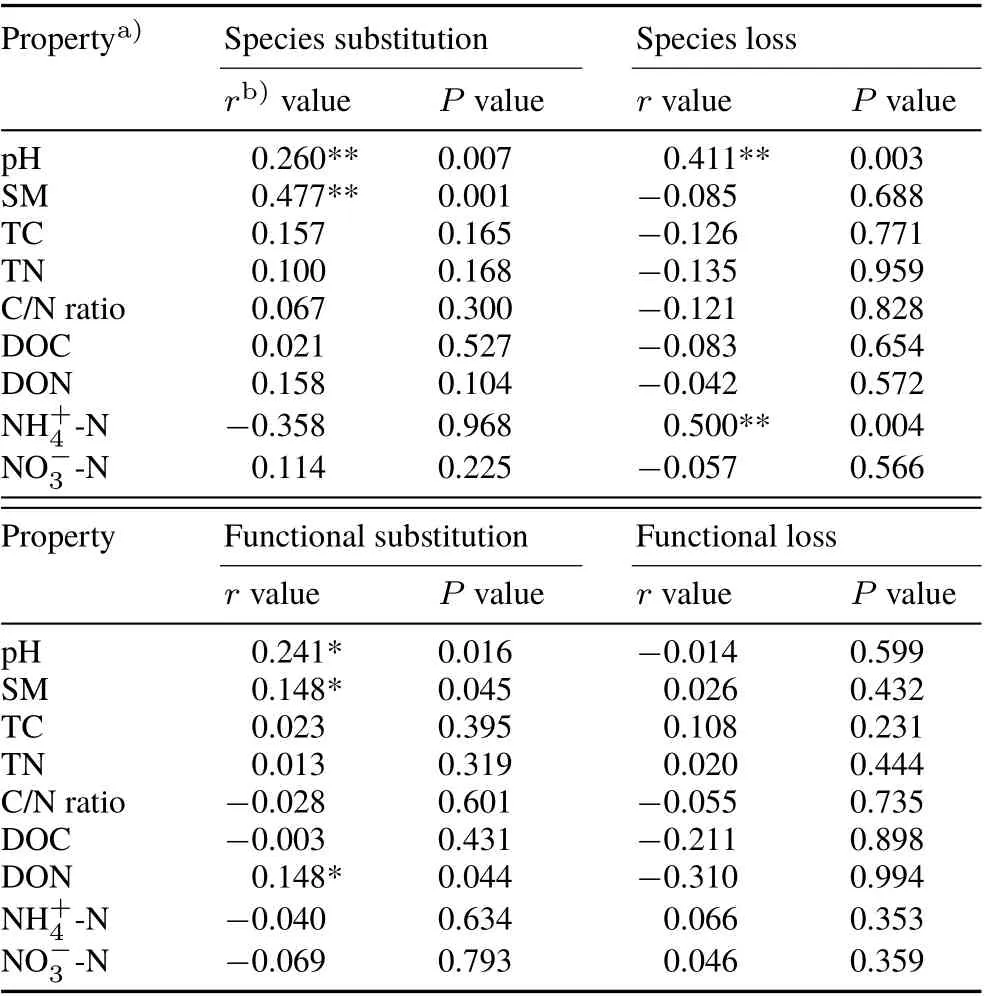
TABLE II Correlations(Mantel tests)between soil properties and species/functional loss and substitution
Bacterial alpha-diversity in the N treatment was significantly lower than in the control and other treatments(Table SI),which was consistent with the general trend where N fertilization decreases the alpha-diversity of a microbial community (Campbellet al., 2010). Bacterial alpha-diversity decreased when the level of N amendment reached 120 kg ha-1year-1in grassland ecosystems in Inner Mongolia,while 60 kg ha-1year-1of N fertilization did not affect bacterial diversity(Zenget al.,2016).In the present study,30 kg ha-1year-1of N application in an alpine wetland ecosystem decreased bacterial alpha-diversity significantly,revealing a high sensitivity of bacterial communities to N deposition.The decrease in bacterial alpha-diversity following N amendment resulted from the loss of unique species(Table SII).Notably,the bacterial alpha-diversity recovered to control level in the WN treatment, suggesting that the effect of N amendment on bacterial alpha-diversity was offset by the lowering of the water table.
Unlike the decreased bacterial alpha-diversity in the N treatment, no significant decrease in functional alphadiversity was observed(Table SIII).Decreases in functional diversity have been observed in an agro-ecosystem(Dinget al., 2018)and a forest ecosystem(Freedmanet al., 2013)following additional N input,which is inconsistent with our results. This inconsistency could be attributed to N input level,since the amount of N fertilizer applied in the agroecosystem was approximately 180 kg ha-1year-1(Dinget al.,2018),while we applied 30 kg ha-1year-1.Microbial functional diversity did not decline until the N input rose to 40 kg ha-1year-1in an alpine meadow ecosystem in the Tibetan Plateau(Zhang and Yuan,2015).Another reason could be the background N deposition level. Greater differences between experimental N input and background N deposition level are more likely to cause significant changes.Following the addition of 30 kg ha-1year-1N input in a northern hardwood forest ecosystem, functional diversity loss occurred only in two study sites with background N deposition levels of 6.8 and 9.1 kg ha-1year-1,but not in two other sites with background N deposition levels of 11.7 and 11.8 kg ha-1year-1(Freedmanet al.,2013).The background N deposition rates in the Tibetan Plateau had reached 13.8 kg ha-1year-1by 2007(Lü and Tian,2007)and would be higher by 2012, which further narrowed the difference between the background N deposition and the artificial N input,resulting in undisturbed functional alpha-diversity.
Soil bacterial and functional beta-diversity
Actinobacteria, beta-Proteobacteria, alpha-Proteobacteria,Bacteroidetes,and Chloroflexi were the most abundant phyla(Table SV).Several mechanisms might be responsible for the changes in these phyla(Tables SVI and SVII).First,N amendment provided more available N in soil,benefiting certain bacteria.Comamonadaceae in beta-Proteobacteria are capable of using N as a denitrifier(Khanet al.,2002).Therefore, greater N amendment could stimulate their growth.Flavobacteria in Bacteroidetes are copiotrophs(Iwaokaet al., 2018; Yanet al., 2018), whose growth could also be stimulated by higher available N.Second,water table lowering introduces an aerobic environment,which is favorable for aerobes. Such groups include Hyphomicrobiaceae in alpha-Proteobacteria,and Planctomycetia in Planctomycetes(DeLonget al.,2014).Conversely,there were decreases in anaerobes such as Anaerolineae in Chloroflexi,Bacteroidia in Bacteroidetes,and Ignavibacteria in Chlorobi.Third,water table lowering led to more alkaline conditions in soil(Table I),leading to significant decreases inPolaromonas glacialisof Comamonadaceae in the W and WN treatments,which were reported to grow at pH 6—7 but not pH 8(Choiet al.,2018).
Soil pH was closely correlated with water table lowering(Table I). Therefore, disentangling the effects of pH and water table lowering on bacterial community statistically is challenging.However,water table lowering was an artificial treatment and should be considered as the primary cause of the subsequent changes, both in soil physicochemical properties and microbial community. For example, water table lowering increased soil pH significantly because more saline ions are precipitated when water is drained from soil.Higher pH affects microbial community.Furthermore,water table lowering could exert its influence by altering soil oxidative stress and cation exchange rates.There were five phyla(alpha-Proteobacteria,Planctomycetes,Chloroflexi,Bacteroidetes, and Chlorobi) responding to more aerobic environment caused by water table lowering and only one phylum(beta-Proteobacteria)responding to increased pH.Therefore,soil pH had direct effects on microbial community,whereas water table lowering effect was indirect and could be exerted further through its influence on various soil properties.
For functional community response, the methanogen functional groups are sensitive to soil water content and O2condition (Boon and Mitchell, 1995). In this study,methanogenesis genes decreased significantly in the W and WN treatments.Alkaline shock genes increased due to an increase in pH from 7.8 in the control and the N treatment to 8.4 in the W and WN treatments(Table I).Lignin degradation genes,which are closely associated with aboveground plant communities(Brown and Chang,2014),were the genes most altered by water table lowering.Aboveground net primary production(ANPP)was significantly lower in a water table lowering treatment in 2011 and did not differ from the control in 2012 (Wanget al., 2017). Since soil microbes exert their ecological functions largely through litter from the last growing season,decreases in lignin degradation genes and other C cycling genes were affected more by ANPP in 2011 than in 2012.In addition,lower water table levels affect genes associated with P cycling by stimulating the release of soil reactive P,leading to an increase in soil available P concentrations(Aldouset al.,2005).
Distinct influence of treatments revealed by beta-diversity partitioning
Bacterial beta-diversity variation can mainly be attributed to species loss under N amendment,while species substitution was dominant in the W and WN treatments(Fig.3).Numerous studies in agricultural and natural ecosystems(Campbellet al.,2010;Ramirezet al.,2010;Zenget al.,2016)have revealed that N fertilization caused a decrease in bacterial alpha-diversity and changes in bacterial betadiversity.We demonstrated that species substitution does not alter beta-diversity under N amendment.A high species loss trend could be ascribed to several factors,including varying dispersal capacity, selective extinction, selective environmental tolerance, or nested habitats (Wrightet al., 1997;Ulrichet al.,2009).In our study,N amendment provided more available N to the nitrophilous bacteria including Comamonadaceae in beta-Proteobacteria and Flavobacteria in Bacteroidetes.Dominance of these groups squeeze the living space for rare species,leading to a decline in overall bacterial richness(Bobbinket al.,2010).Despite insignificant changes in soil pH and ammonium between the N amendment and the control treatments, soil pH and ammonium were correlated with bacterial species loss(Table II).These results were attributable to the cumulative effects that make bacterial community correlate to soil variables,with only minor alterations(Zhanget al.,2016).However,microbial community was able to maintain most of its functional diversity.Therefore,soil microbes that received relatively low N amendments would develop a convergent but functionally stable microbial community, potentially due to strong functional redundancy effects (Rosenfeld, 2002). Similar beta-diversity patterns have been observed in invertebrate communities(Aspinet al.,2018),where seasonal drought caused significant species loss taxonomic communities while maintaining stable functional communities.
Conversely,species substitution could be the mechanism driving community changes following water table lowering,since the alpha-diversity was similar to that of the control.Similarly,beta-diversity differed in a bacterial community,but not alpha-diversity,along a precipitation gradient in a semiarid grassland(McHughet al.,2014)and in an alpine grassland in the Tibetan Plateau(Zhanget al.,2016).Several soil properties,including soil pH,SM,O2limitation,and redox potential,were affected substantially by water table lowering,which imposed selective pressure on a microbial community(Baselga,2010).Here,soil pH and SM were the major edaphic factors linked to microbial species substitution.The disturbances selected new specialists adaptive to the new environment,resulting in a relatively high proportion of specialists(Fig.S1)and the maintenance of total species number (Table SI). Functional community structure also experienced functional substitution,as stress-selected specialists altered the intrinsic functional diversity.However,the new specialists had relatively narrow niche widths(Fig.S1),which indicated that the community was phylogenetically and functionally vulnerable(Aspinet al.,2018).The significant species and functional turnover rates observed in the water table lowering treatments indicated that microbe-mediated C and N allocation could be easily altered,since microbes require C and N resources to facilitate the production of antistress matter and energy(Yerburyet al.,2005).For example,when bacteria experience drought stress, they synthesize more amino compounds as endocellular osmolytes to prevent dehydration,which consumes high C and N resources.Cytoplasmic matter accounts for 40%of the total biomass C and 60%of the total biomass N in stressed bacteria,compared to 3%—6%in stress-free environments(Schimelet al.,2007).Carbon decomposition rate would increase following water table lowering,leading to greater carbon dioxide flux(Burkett and Kusler,2000),shifting wetlands from C sinks to sources,despite decreased methanogenesis rates(Turetskyet al.,2008).Water table levels could also influence N cycling dynamics.Water table lowering triggered intensified N mineralization,resulting in accumulation of ammonium in soil(Chenet al.,2012).This was also observed in our study(Table I). The accelerated mineralization rate could have provided more available N (Freemanet al., 1996), which stimulates microbial nitrification and denitrification(Wanget al.,2017).
CONCLUSIONS
Distinct effects of water table lowering and N amendment on soil microbial phylogenetic and functional communities were demonstrated in an alpine wetland on the Tibetan Plateau.We observed that single N amendment significantly suppressed bacterial diversity.Although bacterial diversity in the water table lowering treatment could recover to the level in the control, the community structure remained largely different.Soil pH and oxidative stress were the major factors that altered dominant bacterial phylum in response to lowered water table.Species loss occurred in the bacterial community,but not in the functional community,due to high functional redundancy. Soil pH and ammonium were the major soil factors driving the processes.Species substitution was the major mechanism that altered both bacterial and functional communities following water table lowering,where soil pH and SM were identified as the major driving factors. The microbial diversity patterns observed under different treatments were insightful and could facilitate the development of biodiversity conservation strategies.Nitrogen deposition has intensified considerably due to human activities such as fuel combustion and agricultural management. Considering that N deposition rates could increase by 50%—100%by 2030 compared to 2000,most likely in South and East Asia (Reayet al., 2008), aboveground plant richness and soil microbial biomass are projected to decrease(Stevenset al.,2004;Zhanget al.,2010).Soil bacterial richness could also decrease owing to soil acidificationviaN deposition(Treseder, 2008). Such scenarios call for urgent efforts to conserve wetlands exposed to high N deposition rates(Reayet al.,2008).On the other hand,water table lowering could alter microbial community structure and intensify C decomposition and N mineralization rates,increasing greenhouse gas emissions.Therefore,stakeholders should put measures into place to conserve original alpine wetland ecosystems in addition to their native microbial communities,which would mitigate microbe-mediated greenhouse gas emissions.
SUPPLEMENTARY MATERIAL
Supplementary material for this article can be found in the online version.
杂志排行

Pedosphere的其它文章
- Letter to the Editor Molecular characterization of an extensively drug-resistant Acinetobacter baumannii isolated from a corn culture soil
- Reclamation of oil-induced soil hydrophobicity in the hyper-arid Evrona Nature Reserve,southern Israel
- Impacts of land use and salinization on soil inorganic and organic carbon in the middle-lower Yellow River Delta
- Rice(Oryza sativa L.)seedlings enriched with zinc or manganese:Their impacts on cadmium accumulation and expression of related genes
- Responses of the methanogenic pathway and fraction of CH4 oxidization in a flooded paddy soil to rice planting
- Effect of biochar applied with plant growth-promoting rhizobacteria(PGPR)on soil microbial community composition and nitrogen utilization in tomato