中草药抑制CYP3A4的效应物质研究进展
2021-12-20张甜甜涂东珠何芋岐葛广波
张 凤,张甜甜,涂东珠,何芋岐,葛广波*
(1.上海中医药大学 交叉科学研究院,上海 201203;2.遵义医科大学 药学院,贵州 遵义 563099)
代谢是大部分药物在体内清除的主要方式,在药物代谢清除的过程中,肝脏中的细胞色素P450酶(CYP/P450)发挥至关重要的作用,在参与药物代谢的P450酶中,CYP3A含量最丰富、底物谱最广,是参与内源性和外源性物质代谢的主要酶[1-2]。作为药物代谢的主要贡献者,CYP3A催化了多种外源物质和内源性化合物(如类固醇)的氧化代谢,因此被认为是调节药物反应和类固醇代谢的关键靶点。CYP3A是人肝脏和人肠中表达最丰富的CYP亚型酶,由4个亚型组成:CYP3A4、CYP3A5、CYP3A7和CYP3A43,位于7号染色体上。其中,CYP3A4和CYP3A5是负责成人药物代谢的主要酶。CYP3A4通常被认为是在人肝中表达的主要形式,而CYP3A5仅存在于约20%的人肝脏中。CYP3A4和CYP3A5在胃、肺、小肠、肾组织中也有表达。CYP3A5与CYP3A4的同源性为83%,其在肝脏中的表达水平低于CYP3A4,但在肾脏中CYP3A5是主要的CYP3A亚型酶[3]。大多数CYP3A4底物也能被CYP3A5代谢。CYP3A7是在人类胚胎,胎儿和新生儿肝脏中检测到的主要CYP亚型酶,在成年肝脏中也检测到,其水平比CYP3A4低得多[3]。CYP3A43是已被报道的人类CYP3A亚家族的最新成员,目前对CYP3A43的研究较少。
许多外源性药物和内源性物质由CYP3A4代谢。内源性物质胆固醇、氢化可的松经CYP3A4代谢被用作CYP3A4酶的活性检测标志物。当前,用于检测CYP3A4酶活性的底物主要有睾酮(Testosterone)、咪达唑仑(Midazolam)、7-苄氧基-4-三氟甲基香豆素[7-benzyloxy-4-(trifluoromethyl)coumarin]、硝苯地平(Nifedipine)、蟾毒灵(Bufalin)、地尔硫卓(Diltiazem)、红霉素(Erythromycin)等。在这些底物中,睾酮和咪达唑仑较为常用,是业界公认的用于检测CYP3A4酶活性的探针底物。由于CYP3A4、CYP3A5和CYP3A7高度的结构同源性,大多数底物没有较好的特异性,因此,大多数底物检测的是CYP3A亚家族的酶活性。
CYP3A参与代谢50%以上的临床常用药物,因此抑制CYP3A可导致药物毒性、中药/药物-药物相互作用和其它不良反应。然而,在某些情况下,抑制/灭活CYP3A也可能对患者带来益处。CYP3A抑制剂的潜在作用如下。
(1)减缓首过代谢,延长药物半衰期。CYP3A抑制剂可通过提高血浆浓度水平来提高快速代谢药物的治疗效率。药物增强原理目前被用于治疗HIV和HCV感染,其中利托那韦及其衍生物洛匹那韦是被用作抗病毒药物的增强剂。该两种上市药物增强剂都是基于CYP3A4晶体结构开发[4-6]。洛匹那韦利托那韦复合片是治疗成人和2岁以上儿童的人类免疫缺陷病毒-1(HIV-1)感染的药物,其中利托那韦是强效CYP3A4抑制剂,洛匹那韦治疗窗宽、代谢较快。在洛匹那韦利托那韦片中,利托那韦通过抑制CYP3A4的活性从而减缓洛匹那韦在人体内的代谢,达到持效作用[7-8]。CYP3A抑制剂具有减缓药物首过代谢,延长药物半衰期的作用。
(2)提升抗肿瘤药物敏感度,逆转多药耐药。很多I相药物代谢酶受孕烷受体(PXR)调控,尤其是PXR是CYP3A基因的外源性物质响应性表达的主要调节剂[9-11]。PXR在人体肝脏和肠道中高表达,且在这些部位中CYP3A分布丰富,能够代谢多种结构多样的内源性和外源性物质[12-14]。很多化疗药物可直接结合到孕烷受体(PXR)的配体结合域上,从而激活CYP3A转录,增加CYP3A酶系的表达,大量的酶代谢了化疗药物而导致耐药[15]。CYP3A的抑制剂可抑制CYP3A的活性,从而减缓抗肿瘤药物在体内的代谢清除,增加抗肿瘤药物的治疗效果,逆转多药耐药。
P-糖蛋白(P-gp)是多药耐药基因(MDR1)的表达产物,CYP3A和P-糖蛋白(P-gp)的底物谱广泛重叠[16],两者共同作用可使药物口服后的首过效应增加,吸收程度降低。另有一些药物是P-gp和CYP3A的共同抑制剂,与P-gp和CYP3A的底物共同服用后能促进底物的吸收[17]。因此,CYP3A抑制剂可以抑制CYP3A的活性,增加抗肿瘤药物在体内的血药浓度,增加抗肿瘤药物的治疗效果,逆转多药耐药。此外,有研究表明CO通过抑制CYP3A4/2C8改变乳腺癌细胞中紫杉醇的药代动力学行为,并确定CO增强了乳腺癌细胞对紫杉醇的敏感性[18]。
(3)调节内源性代谢,用于疾病预防及治疗。多种研究表明,CYP3A4的活性和肿瘤发生发展密切相关[19-22]。Mitra等[20]研究发现,CYP3A4沉默可诱导MCF-7凋亡,抑制Stat3(Tyr-705)磷酸化,而沉默Stat3会阻止乳腺癌细胞生长并消除(±)-14,15-EET(已知影响细胞增殖、迁移和血管生成的代谢产物)诱导的增殖,该研究表明CYP3A抑制剂可以抑制花生四烯酸的代谢,进而抑制癌细胞生长。此外,CYP3A抑制剂可以调节内源性激素代谢,维持体内激素平衡。
(4)中药/药物-药物相互作用早期预警。中药与西药被广泛地结合使用,以治疗多种人类疾病,例如肝脏疾病、心血管疾病和各种类型的癌症[23-24]。CYP3A参与代谢50%以上的临床常用药物,因此多种药物的组合使用可通过抑制CYP3A的活性进而引发不良的中药/药物-药物相互作用或代谢紊乱,尤其是当与一些治疗窗窄的CYP3A底物药物(如华法林、地高辛和某些抗癌药)合用时[24-25]。除此之外,共价结合的CYP3A灭活剂或一些抑制效应极强的CYP3A抑制剂更有可能引发临床相关的中药/药物-药物相互作用[26-27],例如中药苏合香提取物可强效抑制CYP3A介导的睾酮代谢,当与华法林在大鼠体内共同给药时,苏合香延长华法林的半衰期2.30倍,并使华法林的AUC(0-inf)增加了2.74倍[28]。因此,发现CYP3A的抑制剂可提前预测其是否可引发具有临床意义的中药-药物相互作用,为中药-药物相互作用提供早期预警。
当前,多种中药及天然产物被发现可抑制/灭活CYP3A4的活性,包括黄酮类、香豆素类、生物碱类和五环三萜类等化合物[29]。而我们必须要注意的是CYP3A4活性的抑制是一把双刃剑,当CYP3A4抑制剂和某些具有较窄治疗窗的CYP3A4底物药物联合使用时,可能通过抑制CYP3A4引发中药/药物-药物相互作用,导致不良事件或并发症。另一方面,CYP3A4抑制剂可以减缓快速代谢药物的代谢清除来延长药物持效时间,改善具有相对快速清除率的CYP底物药物的治疗结果。因此,发现CYP3A4的抑制剂对于延长快速代谢药物在体内的驻留时间、避免药物毒性等方面均具有指导和借鉴意义。本综述汇总了源于天然的CYP3A4抑制剂,以期为药物临床使用提供指导和借鉴意义。
1 黄酮类对CYP3A4的抑制作用
黄酮类化合物是自然界大量存在的一类天然化合物,包括黄酮、黄酮醇、黄烷酮、黄烷酮醇、异黄酮、异黄烷酮、查尔酮和花青素。它们不仅在一些中草药中含量丰富,而且广泛分布于多种食物中,如水果、蔬菜、谷物、花、茶和葡萄酒等,是人类饮食的重要组成部分[30]。黄酮类化合物具有抗炎、抗氧化、抗过敏、抗癌、抗高血糖和保护心脏等多种生物活性[31]。由于黄酮类化合物具有诸多益处和低毒性,且在我们的日常生活中摄入较高,多项研究已发现黄酮类化合物可调控人肝脏药物代谢酶的活性[32],尤其是CYP3A4。因此黄酮类化合物与其它药物合用的安全性和有效性应在临床上得到更多的关注。
研究表明,多种黄酮类化合物可有效抑制CYP3A4,其中部分黄酮类化合物可时间依赖性抑制CYP3A4。表1中列出了具有CYP3A4抑制活性的部分黄酮类物质[33-57]。Yim 等[35]研究发现中药苦参及其黄酮类化合物对人肝微粒体中多种CYP亚型酶均有抑制作用,其中苦参醇C、苦参醇I、苦参醇M、理查酮A和槐属二氢黄酮G抑制CYP3A4介导的咪达唑仑 1-羟化的半数抑制浓度(IC50)分别为3.95、0.57、1.29、0.69和0.78 μM。进一步的研究表明,苦参醇C、苦参醇I、苦参醇M、理查酮A和槐属二氢黄酮G对CYP3A4的抑制是时间依赖性的,其中苦参醇I对CYP3A4的抑制最强,其灭活CYP3A4的KI和Kinact分别为0.24 μM,0.22 min-1。Li等[45]使用UHPLC-MS/MS鸡尾酒法测定甘草及其14种化合物对9种细胞色素P450酶的抑制作用,发现甘草查尔酮A是一种CYP3A4基于机理的抑制剂,甘草查尔酮A灭活人肝微粒体(HLM)和重组CYP3A4介导的咪达唑仑 1-羟化的KI分别为116.13 μM和2.22 μM。此外,Zhang等[27]发现甘草中多种查尔酮类化合物(甘草查尔酮A、甘草查尔酮B、甘草查尔酮C、刺甘草查尔酮)可时间依赖性抑制CYP3A。Kimura 等[40]研究了60多种多酚类化合物对CYP3A4活性的抑制作用,发现多种多酚类黄酮可强效抑制CYP3A4介导的睾酮 6β羟化,其中穗花杉双黄酮以混合型方式抑制CYP3A4介导的睾酮 6β羟化,其IC50和Ki分别为0.07 μM和0.027 μM。现有结果表明,黄酮类化合物(除黄酮苷类化合物)可强效抑制CYP3A4的活性,双黄酮类化合物对CYP3A4的抑制效应强于单黄酮类化合物。
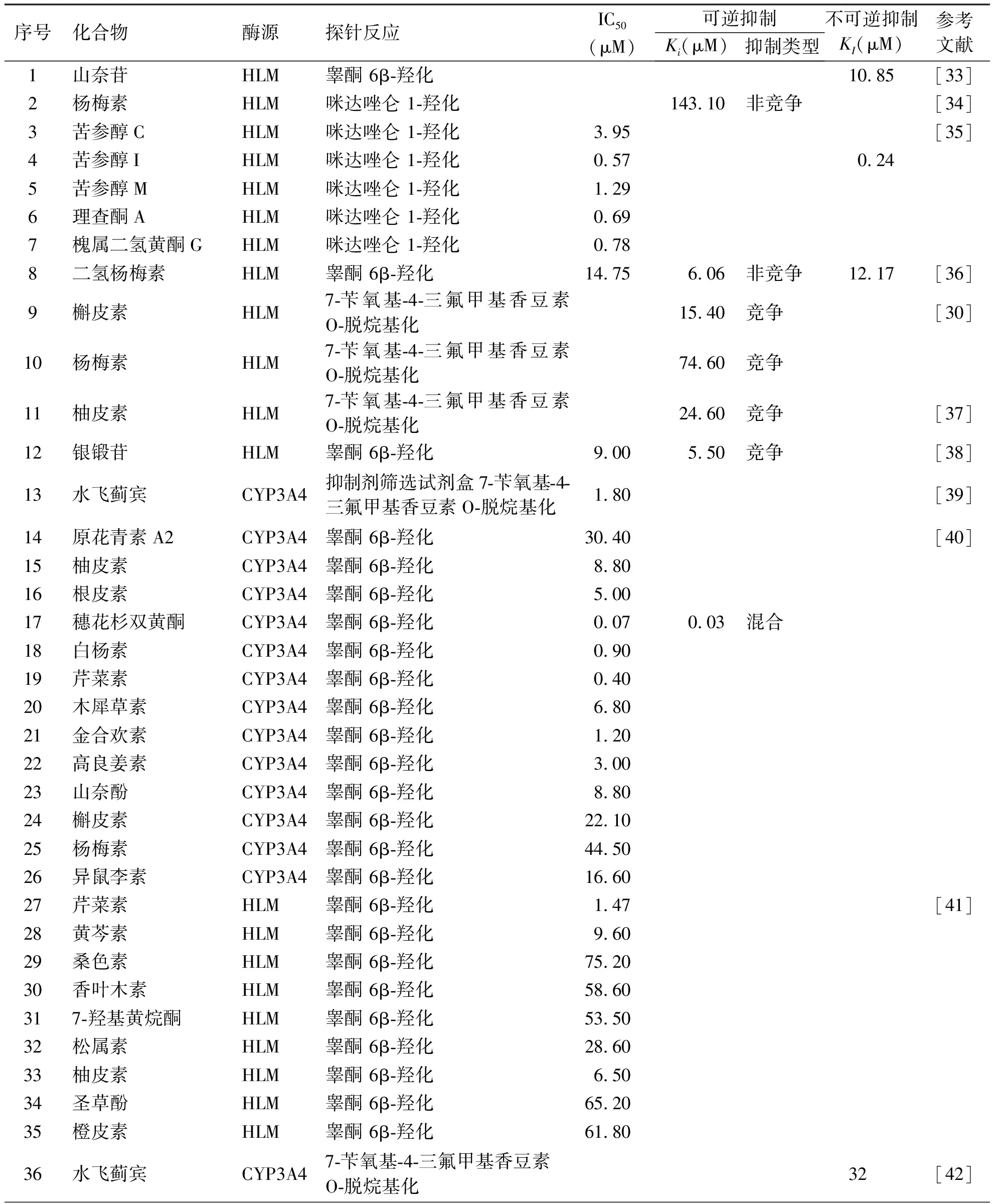
表1 黄酮类化合物对CYP3A4的抑制作用
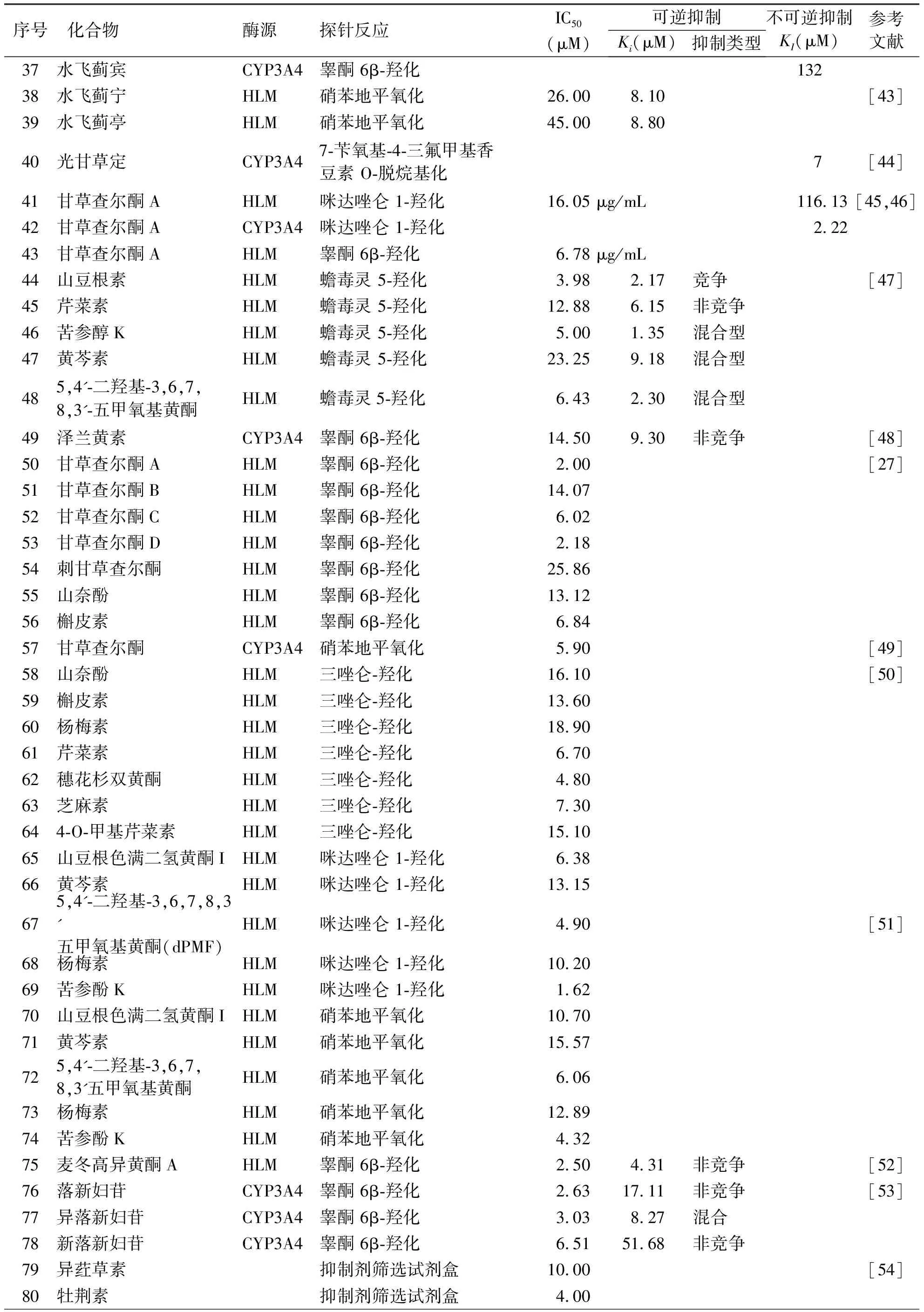
续表

续表
2 香豆素类对CYP3A4的抑制作用
香豆素类化合物是指邻羟基桂皮酸内酯类成分的总称,最基本的结构是苯并α-吡喃酮,广泛分布于高等植物的根、茎、叶、花、果实、皮和种子等各部位,具有抗病毒、抗真菌、抗肿瘤、抗氧化、抗骨质疏松等生物活性[58],是一种重要的天然产物,由于其越来越多的药理活性被发现,引起了天然药物化学家的广泛兴趣,已从药用植物中分离提取了大量的香豆素类化合物。香豆素类化合物的结构类型主要包括简单香豆素类、呋喃香豆素类、吡喃香豆素类和其它香豆素类。
对于香豆素类化合物,目前已发现具有CYP3A4抑制活性的化合物不多,但具有CYP3A4抑制活性的多为呋喃香豆素,且部分呋喃香豆素可时间依赖性地抑制CYP3A4。表2中列出了具有CYP3A4抑制活性的部分香豆素物质[40,59-69]。Song等[60]研究了异嗪皮啶对人肝中CYP3A4的抑制活性,发现异嗪皮啶可抑制CYP3A4的活性,其IC50值为15.49 μM。抑制动力学研究表明,异嗪皮啶是CYP3A4的非竞争性抑制剂,Ki值为10.14 μM。此外,异嗪皮啶还是CYP3A4的时间依赖性抑制剂,其灭活CYP3A4的KI和Kinact值分别为12.33 μM和0.047 min-1。Kimura 等[40]研究了多种香豆素类化合物对CYP3A4活性的抑制作用,发现佛手柑素和欧前胡素(呋喃香豆素)可强效抑制CYP3A4介导的睾酮 6β羟化,其半数抑制浓度分别为0.3 μM和0.5 μM,且欧前胡素以混合型方式抑制CYP3A4,其Ki为0.64 μM,而双香豆素对CYP3A4的抑制效应较弱。
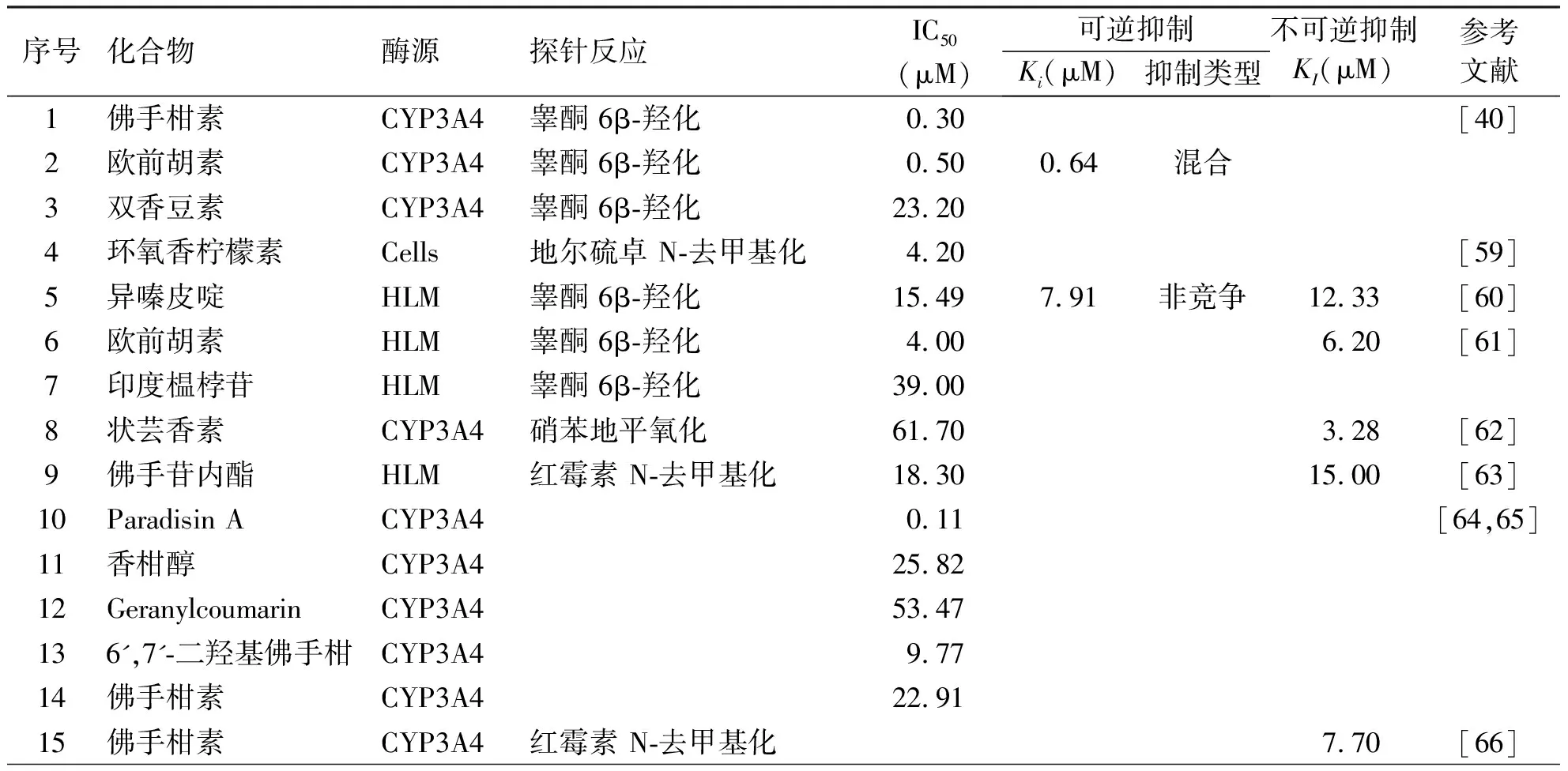
表2 香豆素类化合物对CYP3A4的抑制作用

续表
3 生物碱类对CYP3A4的抑制作用
生物碱类化合物是存在于自然界(主要为植物,但有的也存在于动物)中的一类含氮化合物,是中草药中重要的有效成分之一。多数生物碱类化合物具有抗肿瘤、抗炎镇痛、抗菌等药理活性。表3中列出了具有CYP3A4抑制活性的部分生物碱物质[61,70-82]。Zhang等[70]研究了辣椒碱和二氢辣椒碱对人肝微粒体(HLM)中CYP3A4活性的抑制效应,发现辣椒碱和二氢辣椒碱对CYP3A4介导的咪达唑仑 1-羟化具有中等程度抑制,其半数抑制浓度分别为27.2 μM和61.8 μM;并且辣椒碱和二氢辣椒碱均以非竞争方式抑制CYP3A4介导的咪达唑仑 1-羟化,其Ki分别为32.1 μM和78.6 μM。进一步的研究发现辣椒素对CYP3A4的抑制具有时间依赖性,但作者未测定其灭活常数。Zhao等[74]研究了3种纯化的中药生物碱延胡索乙素,甲基莲心碱和小檗碱对重组人CYP3A4的体外抑制潜力,发现这3种生物碱对CYP3A4的抑制作用较弱,其半数抑制浓度分别为41.5、25.1和48.9 μM。Li等[73]评价了马钱子碱对人肝微粒体中CYP3A4介导的咪达唑仑 1-羟化的抑制作用,发现马钱子碱对CYP3A4的抑制作用较强,其半数抑制浓度为0.77 μM。上述研究表明,生物碱类化合物对CYP3A4的抑制效应普遍较弱,只有较少化合物(例如,马钱子碱)对CYP3A4具有强效的抑制作用。
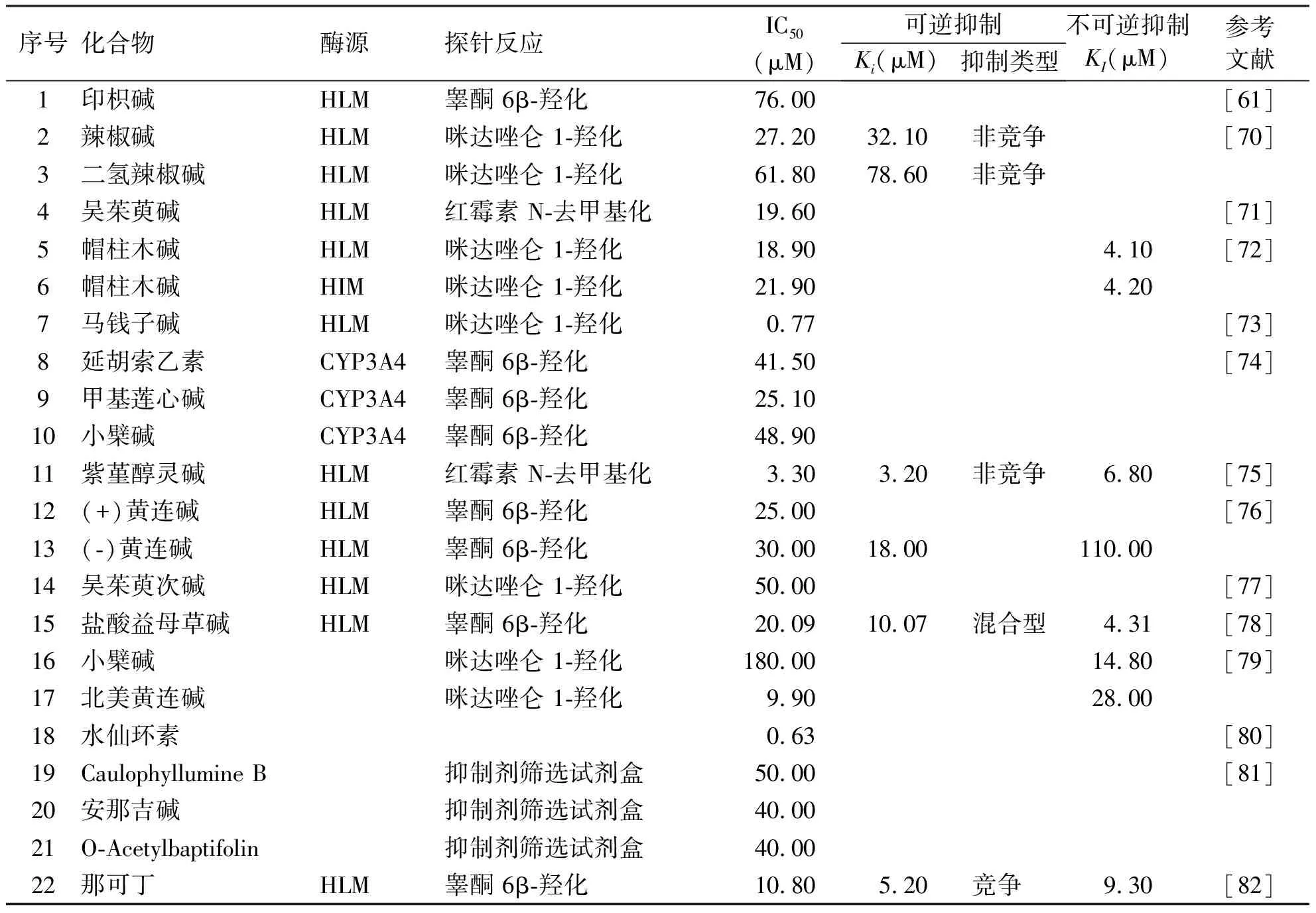
表3 生物碱类化合物对CYP3A4的抑制作用
4 三萜类对CYP3A4的抑制作用
三萜类化合物通常由六个五碳的异戊二烯单元组成,以游离形式或与糖结合成苷存在于药用植物中,多数具有抗肿瘤、抗病毒、抗菌、抗炎和免疫调节等多种药理活性。多种三萜类化合物及其衍生物被证明具有显著和广谱的抗病毒活性。目前已发现的多数三萜类化合物为四环三萜及五环三萜。四环三萜的骨架类型主要有羊毛脂甾烷型、达玛烷型、大戟烷型、原萜烷型、葫芦烷型;五环三萜的骨架类型主要有齐墩果烷型、乌苏烷型、羽扇豆烷型和木栓烷型。
Zhang等[28]从中药苏合香中发现了齐墩果酮酸、白桦脂酸、表白桦脂酸、路路通酸和山楂酸可强效抑制人肝微粒体中CYP3A介导的睾酮 6β-羟化,其半数抑制浓度分别为0.63、4.60、2.87、3.51和4.04 μM。其中齐墩果酮酸可竞争性抑制CYP3A4介导的睾酮 6β-羟化,抑制常数为1.77 μM。进一步的实验研究及分子对接发现,齐墩果酮酸的C-3位的羰基与人CYP3A4的Ser119形成强氢键相互作用,这表明齐墩果酮酸的C-3位羰基可能在与CYP3A4的结合中起关键作用。Sun等[83]的研究发现山楂酸是人体内CYP3A4的竞争性抑制剂。此外,与齐墩果酸相比,山楂酸中C-2位羟基的存在可以增强其对人CYP3A4活性的竞争性抑制作用。Ding等分别发现了茯苓酸和扁塑藤素可抑制人肝微粒体中CYP3A4介导的睾酮 6β-羟化,但是不同的是,他们发现茯苓酸和扁塑藤素对CYP3A4的抑制类型是非竞争[84-85]。也有研究发现熊果酸、科罗索酸、山楂酸可抑制人肠微粒体中CYP3A4介导的咪达唑仑 1-羟化[87]。这些研究表明多数五环三萜类化合物可强效抑制CYP3A4的活性,且可能具有不同的抑制类型(见表4)[84-94]。
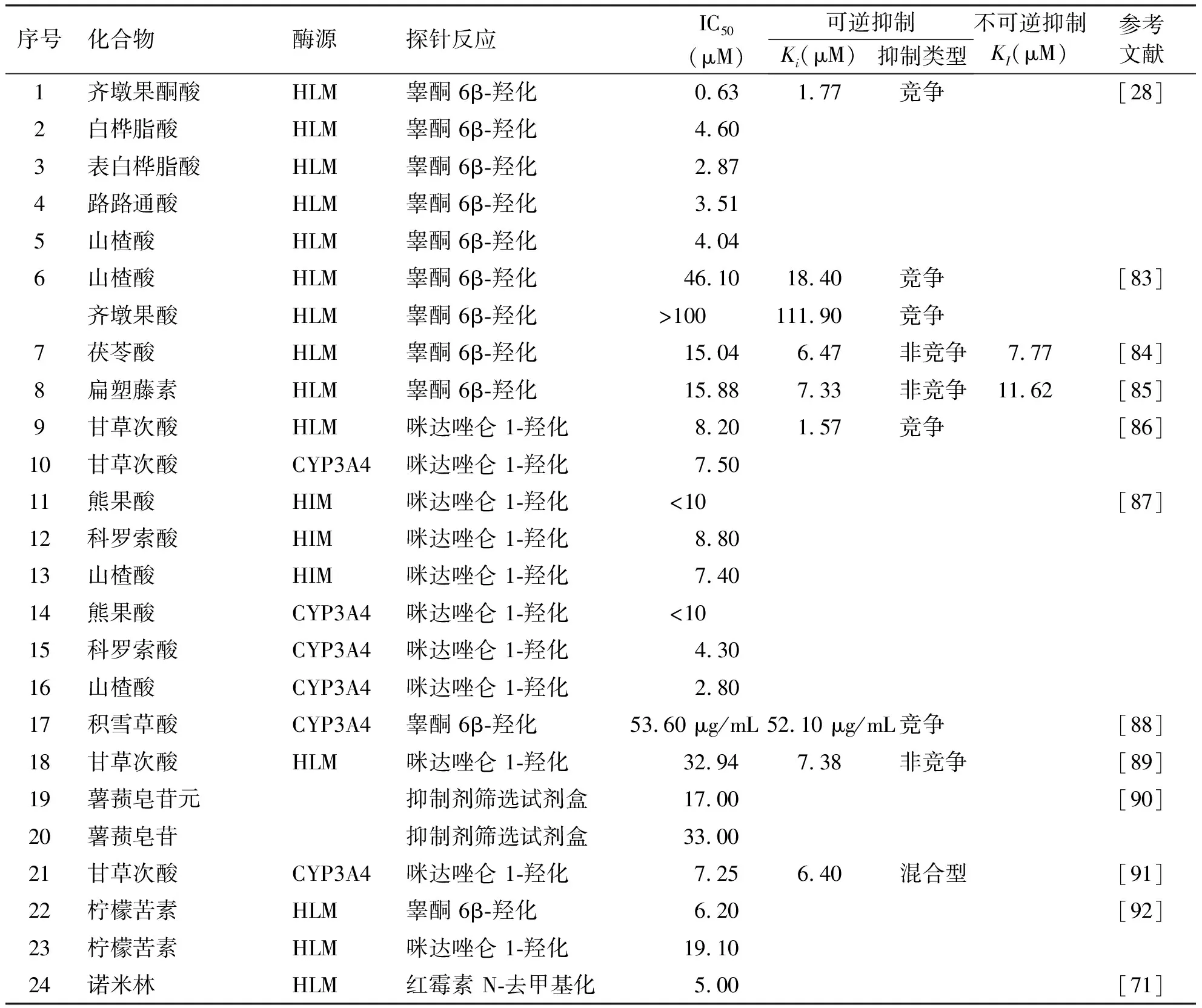
表4 三萜类对CYP3A4的抑制作用
5 有机酸类对CYP3A4的抑制作用
目前,研究发现有机酸类化合物对CYP3A4的抑制作用较弱。Pu等[93]研究了没食子酸对人肝微粒体和重组CYP3A4中睾酮 6β-羟化的抑制效应,发现其抑制CYP3A4的半数抑制浓度均大于100 μM。Pan等[48]研究了迷迭香酸对CYP3A4介导的睾酮 6β-羟化的抑制作用,发现迷迭香酸对CYP3A4的抑制作用较弱,其半数抑制浓度为241.2 μM,且迷迭香酸非竞争性抑制CYP3A4介导的睾酮 6β-羟化,Ki值为328.3 μM。Yao等[94]研究了多种脂肪酸类化合物对CYP3A4介导的咪达唑仑 1-羟化的抑制作用,发现多种脂肪酸类化合物对CYP3A4展示出中等抑制作用,其中亚油酸、亚麻酸、花生四烯酸、二十碳五稀酸、二十二碳六烯酸均竞争性抑制CYP3A4介导的咪达唑仑1-羟化,其半数抑制浓度分别为49、61、48、54和34 μM(见表5)。虽然脂肪酸类化合物对CYP3A4具有中等强度的抑制作用,但人们在日常生活中摄入较多的脂肪酸(包括食用油、肉类等多种食物),因此摄入的脂肪酸是否对CYP3A4具有抑制作用需要视摄入量而定。
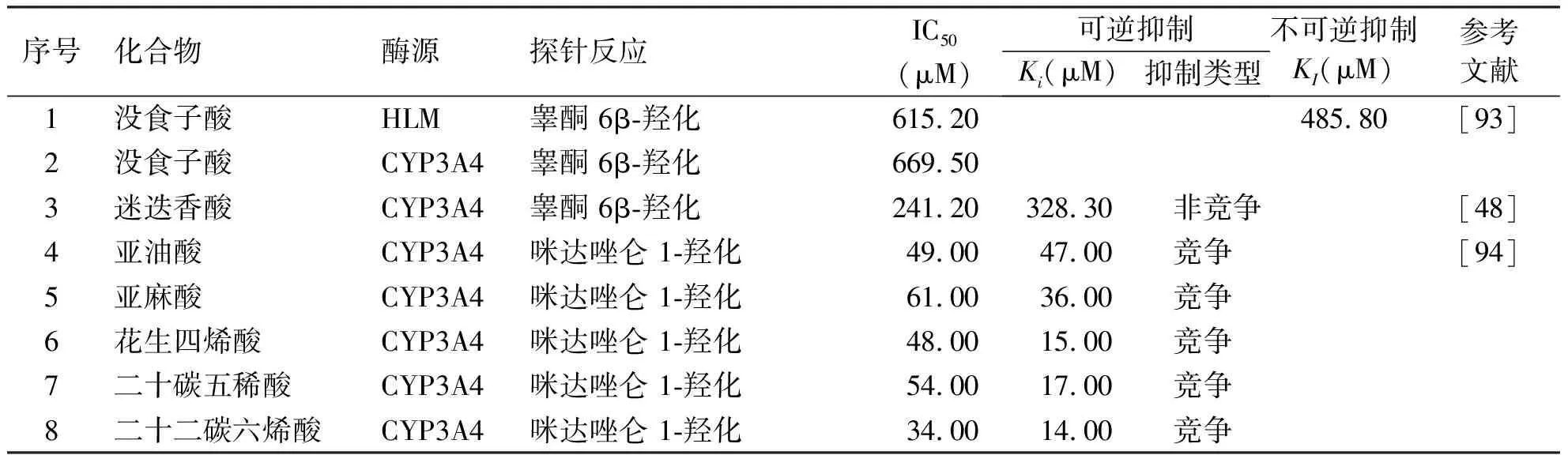
表5 有机酸类对CYP3A4的抑制作用
6 木脂素类对CYP3A4的抑制作用
木脂素是一类由两分子苯丙素衍生物(即C6-C3单体)聚合而成的天然化合物。目前发现的木脂素类CYP3A4抑制剂多来源于中药五味子[97-101]。Iwata 等[98]研究了五味子中的成分对CYP3A4的抑制作用,结果表明,不论使用哪种CYP3A4底物(红霉素和睾酮),五味子醇乙、五味子酯乙、五味子酯甲、戈米辛G和戈米辛N均对CYP3A4有强效的抑制作用。其中五味子醇乙、五味子酯乙、五味子酯甲、戈米辛G和戈米辛N抑制CYP3A4介导的红霉素 N-去甲基化的半数抑制浓度分别为10.1、0.40、0.25、1.02和9.67 μM,且五味子酯甲对CYP3A4的抑制效应最强。五味子醇乙、五味子酯乙、五味子酯甲、戈米辛G和戈米辛N抑制CYP3A4介导的睾酮 6β-羟化的半数抑制浓度分别为6.71、0.62、0.26、1.42和4.32 μM。且五味子酯甲对CYP3A4介导的红霉素 N-去甲基化的抑制是时间依赖性的,其时间依赖性和抑制CYP3A4的KI是0.40 μM(见表6)。
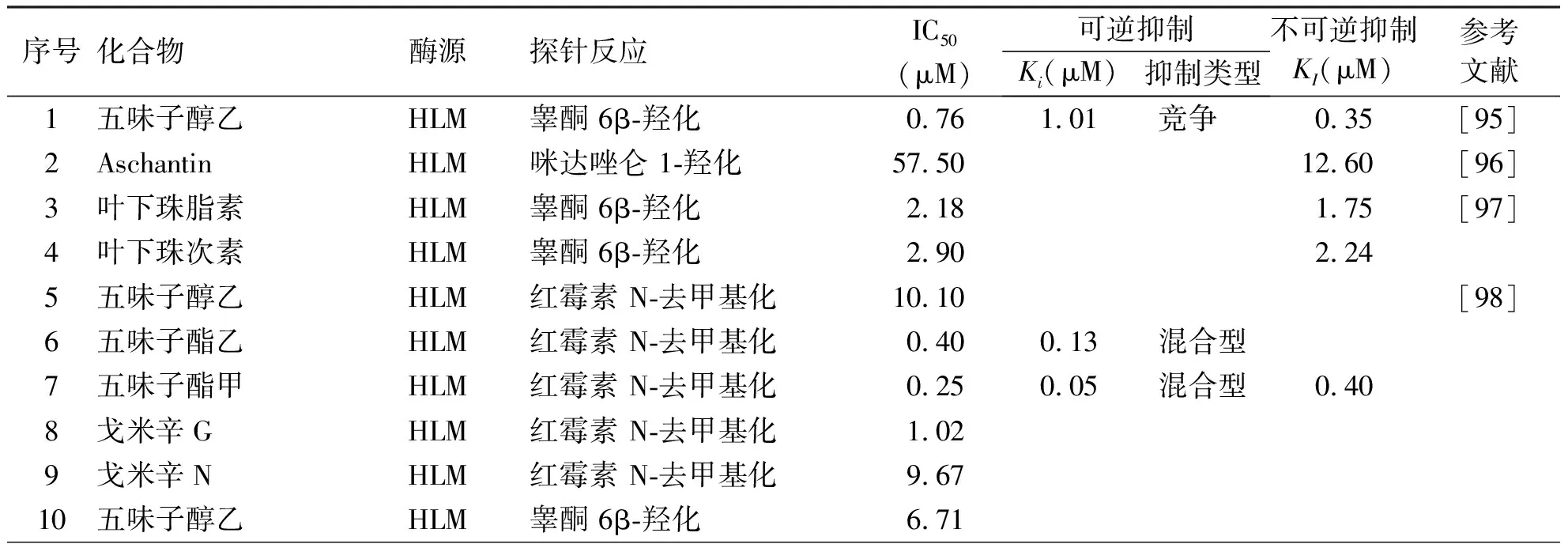
表6 木脂素类对CYP3A4的抑制作用

续表
7 其它化合物对CYP3A4的抑制作用
除黄酮、香豆素、生物碱、三萜、有机酸、木脂类外,还有多种其它类型的化合物被发现可抑制/灭活CYP3A4[102-120]。Gong等[102]发现三白草酮可非竞争性地抑制CYP3A4介导的睾酮 6β-羟化,且抑制CYP3A4的半数抑制浓度(IC50)及Ki值分别为0.21 μM和6.84 μM。Moreira 等[109]研究表明,seriniquinone对CYP3A4介导的咪达唑仑 1-羟化具有强效的抑制作用,其中半数抑制浓度(IC50)为0.12 μM,进一步的研究表明,seriniquinone可时间依赖性灭活CYP3A4,灭活常数(KI)为0.19 μM。另一项研究表明,紫草素可强效抑制CYP3A4介导的咪达唑仑 1-羟化,其半数抑制浓度(IC50)及Ki值分别为2.57 μM和7.72 μM,且抑制类型为混合型[115](见表7)。
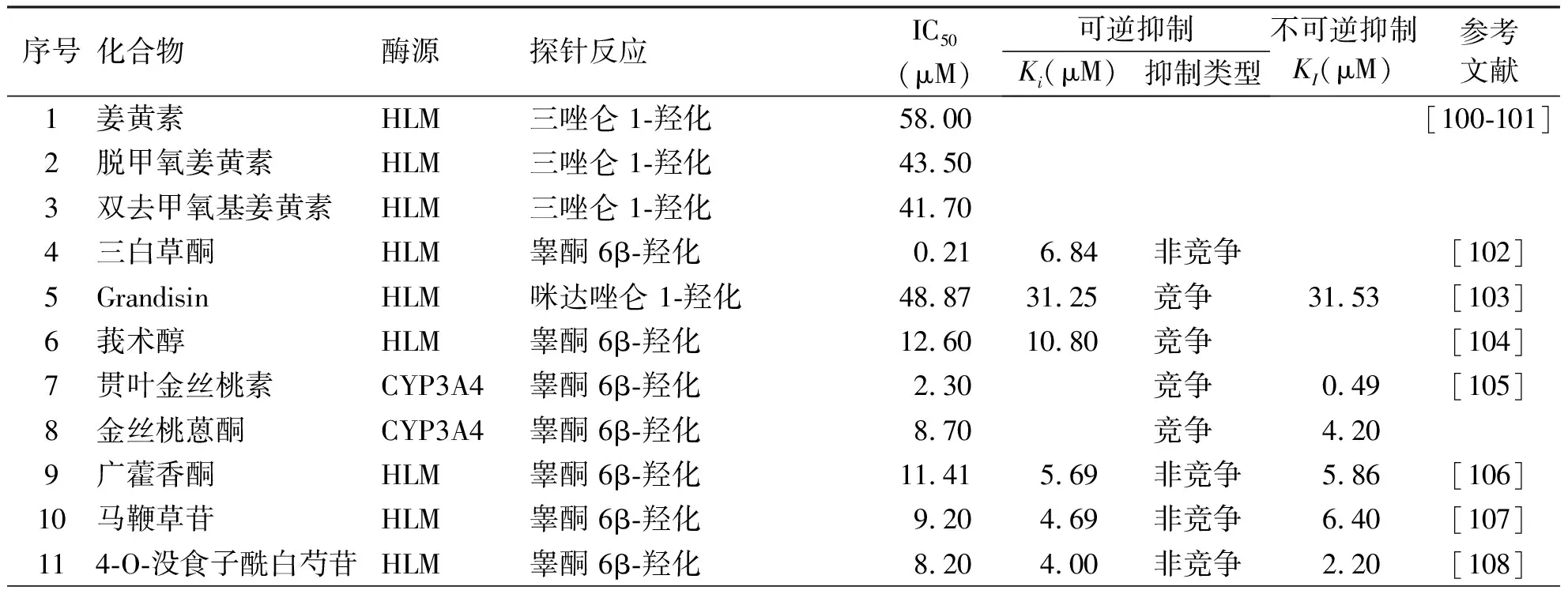
表7 其它化合物对CYP3A4的抑制作用
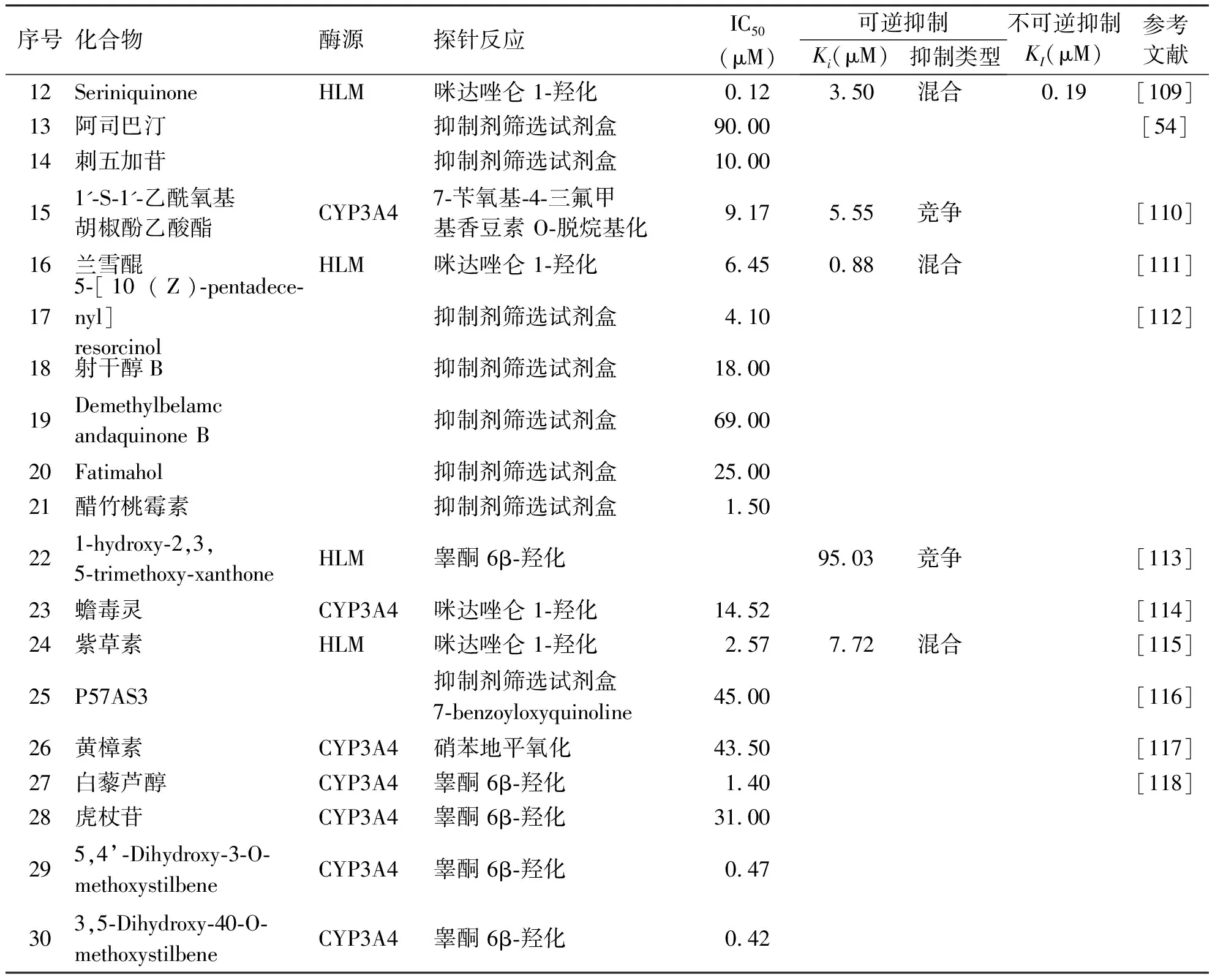
续表
8 总结与展望
CYP3A4是一种含量丰富、底物谱广的药物代谢酶[1-2],目前已上市的药物中有一大部分具有强效的CYP3A4抑制作用,主要包括三类:蛋白酶抑制剂类、大环内酯类抗生素、唑类抗真菌药。随着近年来临床上中西药联用日趋普遍[23-24],并且越来越多的中草药成分被发现可强效抑制/灭活CYP3A4。值得注意的是,共服CYP3A4灭活剂引发的药物不良反应及副作用远高于可逆的CYP3A4抑制剂。为此,本综述着重总结了对CYP3A4具有抑制/灭活作用的中药化学成分,及其抑制/灭活CYP3A4的半数抑制浓度(IC50)、抑制动力学参数(Ki)、抑制类型、灭活动力学参数(KI)等。上述信息有助于指导临床科学合理使用中西药联合疗法,尤其是可避免因CYP3A4灭活引发的中药-药物相互作用及共服化药不良反应的发生。
另一方面,CYP3A4抑制剂可减缓部分底物药物的代谢清除,延长其体内驻留时间和代谢半衰期,提高药物的靶组织暴露量,改善部分可被快速代谢清除的CYP底物药物的治疗结果[5-6,119-121]。如在洛匹那韦利托那韦片中,利托那韦强效抑制CYP3A4延长了底物药物洛匹那韦的代谢半衰期,提高了治疗效果[5-6]。因此,发现安全、特异、强效CYP3A4的抑制剂对于延长快代谢药物在体内的驻留时间、提升药物治疗指数、逆转抗肿瘤药物的耐药性等方面均具有重要意义。但目前已上市的CYP3A4的强效抑制普遍存在特异性差、副作用强、易产生耐药等问题。因此,有必要设计研发更多强效且安全的CYP3A4抑制剂。天然的CYP3A4抑制成分结构类型丰富,可作为筛选新型、安全、高效的CYP3A4抑制剂的来源,但开发过程中需注意从多维度(分子、微粒体、活细胞、活体等)系统评价CYP3A4抑制剂的抑制效能,并同步提升CYP3A4抑制剂的特异性、口服生物利用度、体内代谢半衰期等。
