砂磨制备纳米立方碳化硅及表面特性
2021-12-20邓丽荣王晓刚华小虎陆树河王嘉博王行博
邓丽荣,王晓刚,,华小虎,陆树河,王嘉博,王行博
(1.西安科技大学 材料科学与工程学院,陕西 西安 710054;2.西安博尔新材料有限责任公司,陕西 西安 710089;3.西安科技大学 工程训练中心,陕西 西安 710054)
0 引 言
纳米立方碳化硅由于具有优异的化学稳定性、高温强度、高热导率、高耐磨性和宽禁带、高电场击穿强度等良好特性,在航空、航天、汽车、机械、电子、化工、半导体等工业领域有广泛的应用前景,受到了众多的关注[1-2]。人们在纳米立方碳化硅的制备、微观结构、宏观物性和应用等方面做了大量的研究。如CZOSNEK C以液相有机硅前驱体为原材料,采用气溶胶辅助法对原料进行预处理,再通过直流热等离子体法合成了平均晶粒尺寸从几纳米到几十纳米不等的β-SiC粉体[3]。GUPTA A等采用六甲基二硅烷(HMDS)在电阻加热化学气相沉积(CVD)反应器中合成了SiC纳米粉体,通过改变稀释气体(H2/Ar比)和温度等参数,从结晶度、化学成分和沉积速率等方面对生长条件进行了优化[4]。韩召等以硅粉和炭黑为原料,在N2气氛中通过燃烧合成出纳米SiC粉体[5]。EBADZADEH T等以硅胶和碳为原料,采用常规加热和微波加热两种方法合成了纳米碳化硅粉体[6]。李智敏等采用溶胶-凝胶-碳热还原法,以正硅酸乙酯和蔗糖为原料,在0.1 MPa Ar气气氛中碳热还原合成β-SiC纳米粉体。通过X射线衍射、Raman光谱、扫描电镜和透射电镜对β-SiC纳米粉体的物相、微观结构及形貌进行了表征[7]。虽然纳米立方碳化硅从20世纪90年代就开始发展起来了,但目前仍处于实验研究阶段。常用的纳米碳化硅制备方法如溶胶凝胶法、等离子体法、化学气相沉积法、聚合物热分解法、电弧放电法、自蔓延高温合成法等等[8-13],由于存在技术成熟度不高、制备成本高、产量低、产品稳定性差等问题,目前难以实现大批量工业化生产。目前工业生产采用的碳热还原法虽然具有设备简单、操作容易、生产成本低等优点[13-15],但由于产品粒度大、硬度高,加工成纳米粉体难度较大。如何降低生产成本、扩大应用规模是未来纳米立方碳化硅制备、研究的重点。
随着湿法研磨技术的发展,采用湿法研磨制备纳米粉体是目前最有效且最合乎经济效益的方法。它避免了化学法制备纳米粉体的高成本,也避免了机械干法研磨难以达到纳米级粉体的不足。砂磨机属于湿法超细研磨设备,由于研磨腔狭窄,拨杆间隙小,研磨能量密集,配合高性能的冷却系统和自动控制系统,可实现物料连续加工、连续出料,生产效率极高,是目前物料适应性最广、效率最高的研磨设备,已大量用于纳米涂料、油墨、喷墨、颜料等领域的制备[16-18]。如吴王超等采用机械湿法研磨获得了平均粒径为136.7 nm单颗粒分散的二氧化钛纳米颗粒,并通过正交实验确定其工艺参数[19]。郝嘎子等采用HLG-5型纳米化粉碎机批量制备了粒径约为60 nm的CuCr2O4[20]。但用于高强度高硬度的纳米立方碳化硅的制备鲜见报道。马丽莉利用湿法超细粉碎机只能将碳化硅粒度磨到1.73 μm[21]。
以立方碳化硅为研究对象,采用自主研制的碳化硅介质球作为磨球,在具有一定实验体量的30 L型砂磨机内进行砂磨实验,研究砂磨制备机理,探索批量制备高纯纳米立方碳化硅颗粒的可行性,以及研究机械法制备纳米粉体的表面微观结构变化,以期对纳米立方碳化硅的大批量生产和应用提供一定的理论指导。
1 实验
1.1 实验原料
实验所用原料为西安博尔新材料有限责任公司提供的未经分级处理的立方碳化硅微粉。该微粉采用多热源法固相反应合成,其粒度测试和SEM测试如图1~2所示。从图1可以看出,原料平均粒径Dv(50)为2.49 μm。由于未经分级处理,原料粒径分布从1~6 μm均有,粒度分布稍宽。从图2可以看出,颗粒晶面光滑、未见孔隙,说明原料比较致密。颗粒棱角比较圆滑,多数颗粒为类球形状,小颗粒有轻微团聚现象,粒度分布不太均匀。
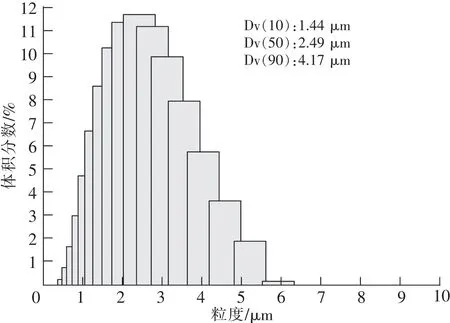
图1 原料粒度测试Fig.1 Volumetric size distribution of raw materials
1.2 实验方法
采用自主研发的粒径为0.3~0.4 mm的碳化硅介质球作为磨球(密度为3.10 g/cm3),去离子水洗净干燥后装填46 kg入SP-30L型砂磨机内。称重50 kg立方碳化硅微粉,加入200 kg去离子水,配成20%固含量的浆料。将浆料置于料缸内搅拌分散后通过隔膜泵打入砂磨机内进行循环砂磨实验。采用该机型最高转速1 484 r/min进行实验,以期获得高的能量输入。实验过程中采用冷水机对筒体进行降温。每隔一段时间进行取样检测。
1.3 测试表征
采用激光粒度仪(英国马尔文MAZ3000)对产物粒度进行测试;采用扫描电子显微镜(德国蔡司Zeiss SIGMA)对产物进行形貌观察;采用高分辨透射电镜(JEM-2800)对产物进行微观形貌和结构测试;采用X射线光电子能谱仪(XPS)(赛默飞世尔科技公司,XPS-Nexsa)对纳米颗粒的表面物质组成进行分析;采用红外光谱分析仪(thermo fisher,Nicolet iS5)进行纳米颗粒表面官能团的测定;采用Zeta电位测试仪(英国马尔文,ZEN3690)进行纳米产物Zeta电位测试和动态光散射粒度测试。
2 结果与讨论
2.1 砂磨机理
机械粉碎法是利用磨机内的磨球与磨球、磨球与磨罐之间的高速高频冲击,使物料受到强烈的冲击、挤压、研磨和剪切等机械力作用而被粉碎成纳米级微粒的方法。图3为不同砂磨时长取样所得的激光粒度测试结果,可以发现在保持工艺参数不变的情况下,碳化硅的粒度特征参数Dv(10)、Dv(50)、Dv(90)在砂磨过程中均经历了一个从快速下降到缓慢下降直至基本不变的过程,变化趋势基本一致。根据各个特征参数的下降速率量级的变化(表1),可将整个砂磨过程分成3个阶段。第1阶段0~10 h,为快速粉碎阶段,碳化硅微粉在磨球的高速碰撞下粒径较大的颗粒发生脆性破坏。由于其晶格缺陷的存在,使得颗粒的实际强度低于理论强度,成为了颗粒粉碎的突破口。在晶界内有气孔、杂质等缺陷的地方产生很大的应力集中,内生裂纹,在外力的冲击下形成沿晶粉碎,从而表现为颗粒的粒径快速下降,颗粒棱角分明。产物的Dv(10)、Dv(50)、Dv(90)相差较大,粒度分布较宽;第2阶段10~30 h,粒径下降速率较第1阶段减小一个数量级,为慢速粉碎阶段。粒径较大的颗粒经过第1次冲击后粉碎成若干小颗粒,颗粒被细化后不再发生脆性破坏,而是发生微塑性变形与疲劳破坏,粉碎效率下降。微小的颗粒主要通过挤压、研磨作用力使其表层剥落,产生不完全粉碎,表现为颗粒的粒径下降速率变缓,Dv(10)、Dv(50)曲线变得平缓,Dv(90)下降缓慢;第3阶段30~40 h,粒径下降速率进一步减小一个数量级,为均化、整形阶段。随着砂磨过程的持续进行,颗粒越细其结构缺陷越小,颗粒强度越高,粉碎难度增加,颗粒粒径趋于稳定,表现为Dv(10)、Dv(50)、Dv(90)基本不变,且相互靠近,反映出粒度分布变窄,颗粒更加均匀。 同时也说明在此工艺下存在砂磨极限,只能制备中位径Dv(50)在100 nm附近的产物(图4)。
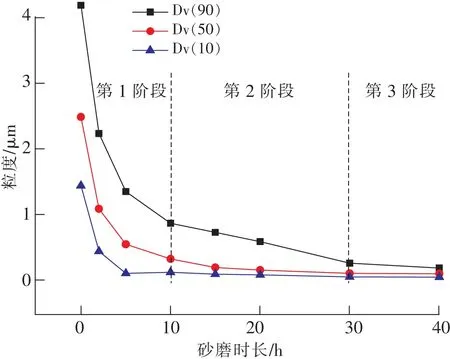
图3 不同砂磨时长取样激光粒度测试Fig.3 Particle size of products with different cumulative grinding time

图4 砂磨粉碎过程Fig.4 Sand grinding process

表1 粒度特征参数的下降速率变化Table 1 Decline rate of each characteristic parameter
2.2 微观形貌
图5为不同砂磨时长取样所得的扫描电镜测试照片。从图5(a)~(c)中可以看出,砂磨第1阶段的产物,颗粒棱角分明、比较尖锐,大颗粒被快速粉碎,大小颗粒夹杂在一起,粒径分布宽;从图5(d)~(f)可以看出,随着砂磨时间的延长,砂磨第2阶段的产物,大颗粒被粉碎成小颗粒,颗粒棱角开始钝化,粒径分布较均匀;而从图5(g)~(h)中可以看到,经过长达30~40 h的砂磨过程,颗粒的尺寸几乎没有变化,但球形度有明显提升,纳米颗粒的粒径分布更加均匀,未见异常大颗粒存在,为颗粒的均化、整形阶段。所得的扫描电镜测试结果也为上述的砂磨机理分析提供了更为直观的证明。

图5 不同砂磨时长取样扫描电镜测试Fig.5 SEM of SiC powders with different cumulative grinding time
采用透射电镜对砂磨纳米产物进行微观形貌分析,结果如图6所示。从图6(a)可以看出,所得砂磨纳米产物主要由一种近似球形的纳米颗粒组成,颗粒分布比较均匀,颗粒尺寸在30~50 nm之间,粒径分布较窄,且颗粒的球形度较好。图6(b)电子衍射花样为一系列不同半径的同心圆环组成,为选区内大量取向不一的细小晶粒产生的,说明纳米产物为多晶相。

图6 纳米碳化硅颗粒TEMFig.6 TEM of SiC nanoparticles
2.3 表面结构
纳米颗粒的表面特性包括组成和结构,对纳米颗粒的分散及应用非常关键。采用X射线光电子能谱仪和傅里叶红外光谱仪对砂磨纳米产物的表面成分和表面官能团进行测试,结果如图7和图8所示。图7为砂磨得到的纳米立方碳化硅的XPS拟合分析谱图,在结合能101 eV附近对应的是Si—C键,在结合能103 eV附近对应的是Si—O2键,结果显示纳米立方碳化硅颗粒表面除了SiC成分还有含量较高的SiO2出现[22]。颗粒表面所形成的SiO2为砂磨过程中由于粒度减小,比表面积增大,纳米颗粒在强大的机械力冲击下表面发生氧化所致。
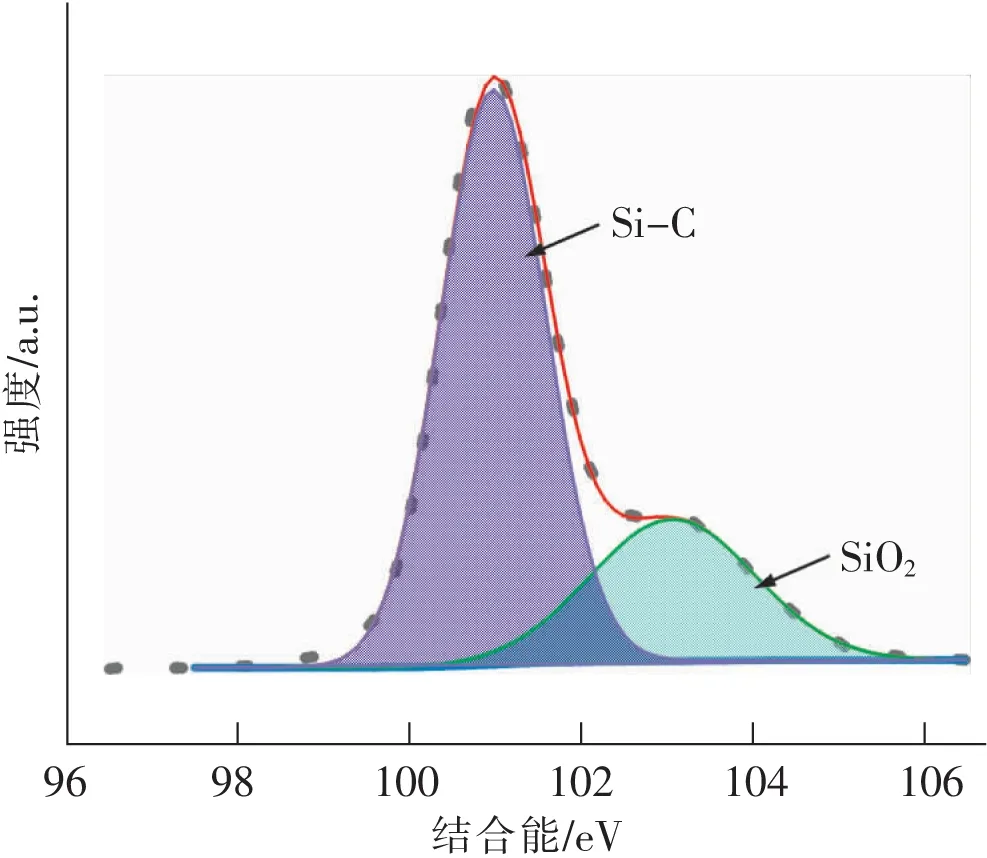
图7 纳米立方碳化硅的XPS拟合Fig.7 XPS fitting of nano cubic silicon carbide
图8为砂磨原料与砂磨纳米产物的红外光谱图。对比图8(a)和(b)可以发现,砂磨纳米产物在3 407.4 cm-1处表现出宽峰强吸收,是由于O—H键的伸缩振动引起的。同时在1 623.9 cm-1附近为水的H—O—H弯曲振动峰,1 072.2 cm-1附近的吸收峰为Si—O—Si 反对称伸缩振动峰,462.8 cm-1的峰属于Si—OH的弯曲振动峰。这说明纳米立方碳化硅表面覆盖了大量的羟基[23],亲水性强,且含有SiO2官能团,与XPS的检测结果一致。
2.4 纳米浆料分散稳定性
DLVO理论认为,分散体系的稳定性是当颗粒相互接近时它们之间的扩散双电层互斥力与粒子间范德华力相互吸引的净结果。在立方碳化硅分散体系中,碳化硅颗粒会从周围的溶液中有选择性地吸附离子,使其表面带电荷。颗粒表面和周围溶液的电势差可通过Zeta电位进行测试。Zeta电位绝对值越大,颗粒之间的排斥力越大,分散体系越稳定。若Zeta电位为零,在此等电点附近,则体系的分散稳定性较差,颗粒之间易团聚形成大颗粒而发生沉降[24]。在不同的pH值条件下,砂磨所得纳米浆料的Zeta电位,测试结果如图9所示。从图10可以看出,与化学法合成的纳米立方碳化硅的等电点在pH在3附近不同[8,25],砂磨制得的纳米立方碳化硅浆料在测试范围内没有出现等电点。结合前面的红外光谱测试和XPS测试分析结果,分析可能是纳米立方碳化硅在砂磨过程中由于受到持续的机械力作用,颗粒表面形成了无定形的二氧化硅氧化层[25],使得纳米浆料的表面特性与二氧化硅相近,测试结果与Sakthivel等报道的二氧化硅的Zeta电位测试结果也大致相同[26]。纳米立方碳化硅浆料在pH≥7,其Zeta电位绝对值都超过40 mv,处于分散稳定性较好水平,尤其在pH为9附近,Zeta电位绝对值达到60 mV以上,分散稳定性极好。
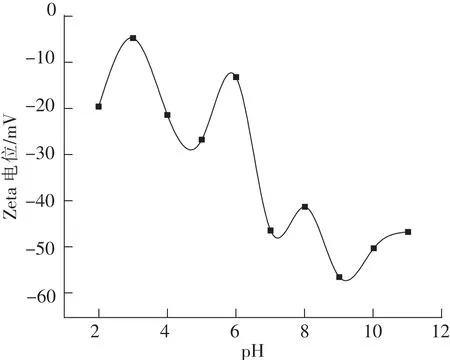
图9 纳米碳化硅Zeta电位与pH值的关系Fig.9 Relationship between Zeta potential and pH value of nano SiC

图10 不同pH值下纳米颗粒的粒径Fig.10 Particle sizes of nanoparticles at different pH values
采用动态光散射对不同pH值下的纳米颗粒进行粒度测试,其结果如图10所示。从图10可以看出,不同pH值下纳米碳化硅的粒度测试结果变化较大。在酸性环境下,尤其是pH<5时,纳米颗粒的分散稳定性比较差,颗粒团聚比较严重,形成的团聚颗粒最大可达620 nm;而在中性和碱性环境下,pH值≥7后,纳米颗粒的分散稳定性较好,其粒径均小于200 nm,尤其在pH在9~10附近时,纳米颗粒粒径测试达到最小值103 nm,分散稳定性最好。粒度测试结果与Zeta电位测试结果相一致。表明砂磨制备的纳米碳化硅颗粒在中性和碱性条件下分散稳定性比较好,可用于机械抛光[27]、陶瓷湿法成型、涂层等工业应用领域。
3 结 论
1)以平均粒径Dv(50)为2.49 μm立方碳化硅为原料,采用0.3~0.4 mm的碳化硅介质球,经砂磨40 h可批量制备Dv(50)在100 nm附近的纳米颗粒。
2)砂磨过程中产物的粒径下降速率会发生数量级的变化,可将其划分为快速粉碎、慢速粉碎和均化、整形3个阶段。
3)SEM和TEM测试结果表明,砂磨制备的纳米立方碳化硅颗粒,单颗粒尺寸在30~50 nm之间,粒径分布较窄,且颗粒的球形度较好。
4)砂磨制备的纳米立方碳化硅,表面含有大量的羟基和二氧化硅,具有很好的亲水性。其表面特性与纳米二氧化硅相近。在pH≥7下分散稳定性良好,尤其在pH为9时Zeta电位绝对值最高可达60 mV以上,粒径达到最小值103 nm,分散稳定性最好,可用于机械抛光、陶瓷湿法成型、涂层等工业应用领域。
