核外p53通过AMPK/mTOR信号抑制自噬并促进热打击诱导的血管内皮细胞损伤
2021-12-16邹志敏古正涛
李 莉,邹志敏,李 琴,张 堃,苏 磊,古正涛
1南方医科大学第三附属医院创伤救治中心,广东 广州 510630;2广东省骨科研究院//广东省骨科医院//广东省骨与关节退行性疾病重点实验室,广东 广州 510630;3中国人民解放军南部战区总医院重症医学科,广东 广州510010
我们前期实验结果发现,在热打击的血管内皮细胞(ⅤECs)模型中,核外p53(即p53的线粒体移位)介导了细胞线粒体损伤、线粒体膜透化,最终诱导ⅤECs广泛损伤[1,2];核外p53主要是通过抑制自噬而导致了ⅤECs的广泛损伤[3],但这一过程中的具体机制仍不明确。既往研究提示,腺苷酸活化的蛋白激酶(AMPΚ)/雷帕霉素靶体(mTOR)信号通路参与调控自噬过程,并且p53可以通过磷酸化AMPΚ调控自噬抑制因子mTOR活性而发挥自噬促进/抑制功能[3-5]。然而,热损伤过程中核外p53是否通过AMPΚ/mTOR介导细胞自噬抑制参与血管内皮细胞损伤,目前为止国内外尚无相关报道。基于以上的发现和认识,我们推测:在热打击ⅤECs中,核外p53通过抑制AMPΚ活性,激活mTOR信号,继而介导细胞自噬抑制,而细胞自噬不足最终诱导ⅤECs出现广泛损伤,从而阐明热打击后ⅤECs损伤的分子机制。
1 材料和方法
1.1 实验动物
SPF级,C57BL/6小鼠,雄性,6~8周龄,体质量22~25 g,由南方医科大学实验动物中心提供,许可证号:SCXΚ(粤)2016-0167。所有饲养及实验皆依照《南方医科大学实验动物管理办法(试行)》和《南方医科大学动物实验伦理审查指南(试行)》原则进行。
1.2 实验仪器
多功能酶标仪(Molecular Devices);Image J图像工作站(Κodak);超净工作台(中国);二氧化碳孵箱(SANYO);低温离心机(Allegra X-22R,BECΚMAN COULTER);转膜仪(Amersham Phamacia Biotech);凝胶成分析仪(ⅤilberLounnat);数据处理系统(SANYO,日本);图像分析(SANYO);倒置相差显微镜(Leica,SP2 AOBS);激光共聚焦扫描显微镜(Leica,SP2 AOBS);流式细胞仪(Becton Dickinson,FACSCalibur)。
1.3 实验试剂
DMEM高糖培养基、胎牛血清、胰酶、双抗(Gibco);BCA蛋白定量试剂盒;组织、细胞总蛋白蛋白提取试剂盒、胞质和线粒体分离提取试剂盒(贝博);CCΚ-8细胞计数试剂盒(碧云天);0.2 μm 转印膜(Millipore);Pifithrin-a(PFT)(Calbiochem Merk);Compound C 和rapamycin(Sigma);p53、LC3-Ⅱ、Beclin-1、P62、p-AMPΚ、AMPΚ、p-mTOR、mTOR、p-4EBP1、4EBP1、p-p70S6Κ、p70S6Κ等抗体(Abcam);GAPDH抗体、辣根过氧化物酶标记山羊抗小鼠IgG(H+L)、辣根过氧化物酶标记山羊抗兔IgG(H+L)以及荧光二抗(碧云天)。
1.4 主动脉内皮细胞(MAECs)原代分离和鉴定
取6~8周龄,体质量22~25 g的C57BL/6小鼠,腹腔注射肝素钠1250 U,20 min后用7.5%的水合氯醛腹腔注射麻醉(约11.25 g/kg)。参照课题组前期方法进行[1],麻醉下无菌操作,完整分离动脉,动脉血管腔内注入2 mg/mL Ⅱ型胶原酶,37 ℃、5%CO2细胞培养箱中孵育45 min,收集血管腔内溶液,离心后接种于涂布Ⅰ型胶原酶基质的35 mm培养皿,并置入37 ℃、5%CO2细胞培养箱中孵育。分离好的内皮细胞,甲醇固定,5%BSA室温封闭1 h,孵育ⅤE-Cadherin和α-actin抗体(1∶200),室温2 h,预冷磷酸缓冲液(PBS)漂洗3次(2 min/次),加入荧光二抗(1∶500),室温避光孵育1 h,20 μL含猝灭剂及hoechest33258混合液(1∶1),避光孵育10 min,激光共聚焦显微镜拍照。加入vWF抗体(1∶200),4 ℃过夜,加入辣根过氧化酶标记的anti-rabbit二抗(1∶200),37 ℃反应60 min。加入DAB反应液显色,观察阳性染色的细胞比例。
1.5 建立热打击体外模型
参照前期实验方法建立细胞热打击模型[1,6],即实验前1 d MAECs按1.0×105/mL密度将细胞铺于培养皿中,实验分组:对照组(37 ℃)、热打击组(43 ℃)、热打击组+Compound C 组、热打击组+rapamycin 组、热打击组+PFT组。对照组细胞始终置于37 ℃、5%CO2细胞培养箱中。热打击组:封口膜密封细胞培养瓶口后,置于恒温水浴箱中,细胞培养瓶保持液面距离5 cm(同一位置放置温度计监测温度),水浴箱温度维持于43±0.5 ℃,热打击时间为2 h,热打击后将细胞置于37 ℃、5%CO2细胞培养箱中按不同时间点(0、3、6、9 h)进行复温;热打击组+Compound C 组、热打击组+rapamycin组、热打击组+PFT 组,分别使用AMPΚ 抑制剂Compound C(5 μmol/L)[7]、mTOR抑制剂(自噬激活剂)rapamycin(20 μmol/L)[8]、p53 线粒体转位抑制剂PFT(10 μmol/L)[3]预处理细胞1 h。
1.6 建立热打击体内模型
参照前期实验方法建立细胞热打击模型[1,6],实验分组:对照组(37 ℃)、热打击组(43 ℃)、热打击组+Compound C 组、热打击组+rapamycin 组、热打击组+PFT组。各组动物在常温25.0±0.5 ℃下恢复6 h。常温对照组小鼠始终置于常温下;高热刺激组小鼠置于仿真气候舱内,舱内温度39.5±0.5 ℃,湿度(60±5)%,肛温表监测小鼠直肠温度,15 min/次。重症中暑诊断标准:核心体温达到43 ℃维持时间超过1 min。达到重症中暑诊断标准后,小鼠移至常温复温6 h后分离主动脉血管。
1.7 细胞存活率测定
取原代培养状态良好的MAECs,对照组和热打击组分别设置3个复孔,分别加入CCΚ8 10 μL/孔,操作参考说明书。酶联免疫检测吸光度值A450nm,计算细胞存活率。实验分别独立重复3次。
1.8 Western blot检测目的蛋白的表达情况
收集对照组与热打击组的MAECs,按细胞组分分离试剂盒说明书提取线粒体与细胞质。BCA法定量后,SDS聚丙烯酰胺凝胶电泳后,将蛋白转移至硝酸纤维素膜,5%BSA温封闭2 h,洗膜后加入p53、LC3-Ⅱ、Beclin-1、p62、p-AMPΚ、AMPΚ、p-mTOR、mTOR、p-4EBP1、4EBP1、p-p70S6Κ、p70S6Κ 等一抗(1∶1000)4 ℃过夜,TBST洗3遍后,加入二抗(1∶5000)室温孵育2 h后使用化学发光剂ECL进行反应、曝光。
1.9 免疫荧光观察目的蛋白的表达情况
取原代培养状态良好的MAECs进行热打击,多聚甲醛固定,PBS漂洗,BSA封闭,加入LC3-Ⅱ一抗,4 ℃孵育过夜;加入FITC 绿光标记二抗,37 ℃孵育1 h,PBS漂洗,封片,激光共聚焦显微镜下观察。
1.10 HE染色
取各组主动脉血管进行固定、石蜡包埋,切片后行HE染色,主要步骤如下:切片加入苏木素染液约8 min,自来水冲洗15 min,室温下盐酸酒精分化30 s,自来水冲洗至返蓝,伊红染液复染30 s,自来水冲洗,梯度酒精脱水、二甲苯透明,中性树胶封片,光镜下观察比较各组动物主动脉内皮血管病理学变化。
1.11 TUNEL法检测组织凋亡
将石蜡包埋的组织切片置于染色盒中,将洗涤后的组织切片中加入含2%过氧化氢的PBS,室温孵育5 min,PBS 洗5 min×2 次,滴加适量的TdT 酶缓冲液,37 ℃湿盒中反应1 h,加入37 ℃洗涤与终止反应的缓冲液30 min,PBS洗5 min×3次,加100 μL HRP 30 min,PBS洗5 min×4 次,加100 μLDAB混合液15 min,洗涤5 min×4 次,苏木素复染,自来水冲洗、梯度酒精脱水、二甲苯透明、中性树胶封片,显微镜下观察组织中凋亡情况。
1.12 统计学方法
采用SPSS 19.0统计软件包进行分析。计量资料,在方差齐性的基础上应用单因素方差分析,组间差异用LSD法比较,P<0.05为差异具有统计学意义。
2 结果
2.1 热打击诱导MAECs细胞活力,p53线粒体移位
首先对原代的小鼠主动脉内皮细胞(MAECs)进行鉴定,可见MAECs边界清楚,呈类圆形、多角形以及梭形等,胞浆丰富,细胞大小均匀,排列紧密,互不重叠,呈现内皮细胞“鹅卵石”样特征(图1 A-a);使用vWF免疫细胞化学染色观察发现,MAECs胞浆染成棕黄色,且阳性细胞数达95%以上(图1A-b);并使用ⅤE-Cadherin对MAECs进行荧光染色,激光共聚焦显微镜观察发现细胞间边缘连接处的红色荧光线性结构为ⅤE-Cadherin(图1 A-c);使用平滑肌细胞标志物α-actin对MAECs进行荧光染色,激光共聚焦显微镜下未检测到α-actin荧光信号(图1A-d)。
进一步实验检测热打击对MAECs损伤的影响,我们观察了43 ℃热打击2 h后不同复温时间点(0、3、6、9 h)MAECs的细胞活力改变情况,结果显示,热打击后MAECs的细胞活力呈复温时间依赖性下降(图1 B,P<0.05);同时,分别提取MAECs线粒体和胞质蛋白进行Western blot分析,发现热打击后MAECs线粒体中p53随着复温时间延长不断增多,而胞质中p53随着复温时间延长不断减少(图1C)。

图1 热打击后细胞活力下降,p53线粒体移位Fig.1 Heat stress (HS) induces cell viability reduction and p53 translocation from the cytoplasm to mitochondria in mouse aortic endothelial cells(MAECs).A:Isolation and identification of MAECs by morphological observation under optical microscope(Original magnification:×100;a),immunocytochemical staining of vWF(×100;b),and fluorescence staining of VE-Cadherin (red fluorescence;c) and α-actin (red fluorescence;d) observed under confocal laser scanning microscope.B:Cell viability was detected by CCK-8 assay.C:Levels of p53(mitochondria and cytosolic fraction)proteins detected with Western blotting.*P<0.05 vs control group.
2.2 热打击诱导MAECs 中自噬相关蛋白LC3-Ⅱ、Beclin-1和P62以及AMPΚ/mTOR信号通路的改变
Western blot检测结果显示LC3-Ⅱ在热打击后即开始表达,3 h达顶峰,6 h显著下降(图2 A、B,P<0.05),Beclin-1蛋白表达与LC3-Ⅱ蛋白表达的趋势呈一致性(图2 A、C,P<0.05);而p62蛋白在热打击后被抑制,6 h后开始激活,与LC3-Ⅱ和Beclin-1蛋白表达呈现相反趋势(图2 A、D,P<0.05)。热打击后早期即引起MAECs中自噬的激活,但随着复温时间的延长导致了热打击诱导的自噬逐渐被抑制。
进一步实验观察热打击后MAECs 中AMPΚ/mTOR信号通路的变化情况,结果发现热打击后即诱导了AMPΚ的磷酸化激活,3 h达高峰,6 h后被抑制(图2E、F,P<0.05);mTOR 以及其下游蛋白4EBP1 和p70S6Κ的磷酸化在热打击后明显下调,6 h后表达开始上调,与AMPΚ的磷酸化呈现相反趋势(图2B、G~I,P<0.05)。
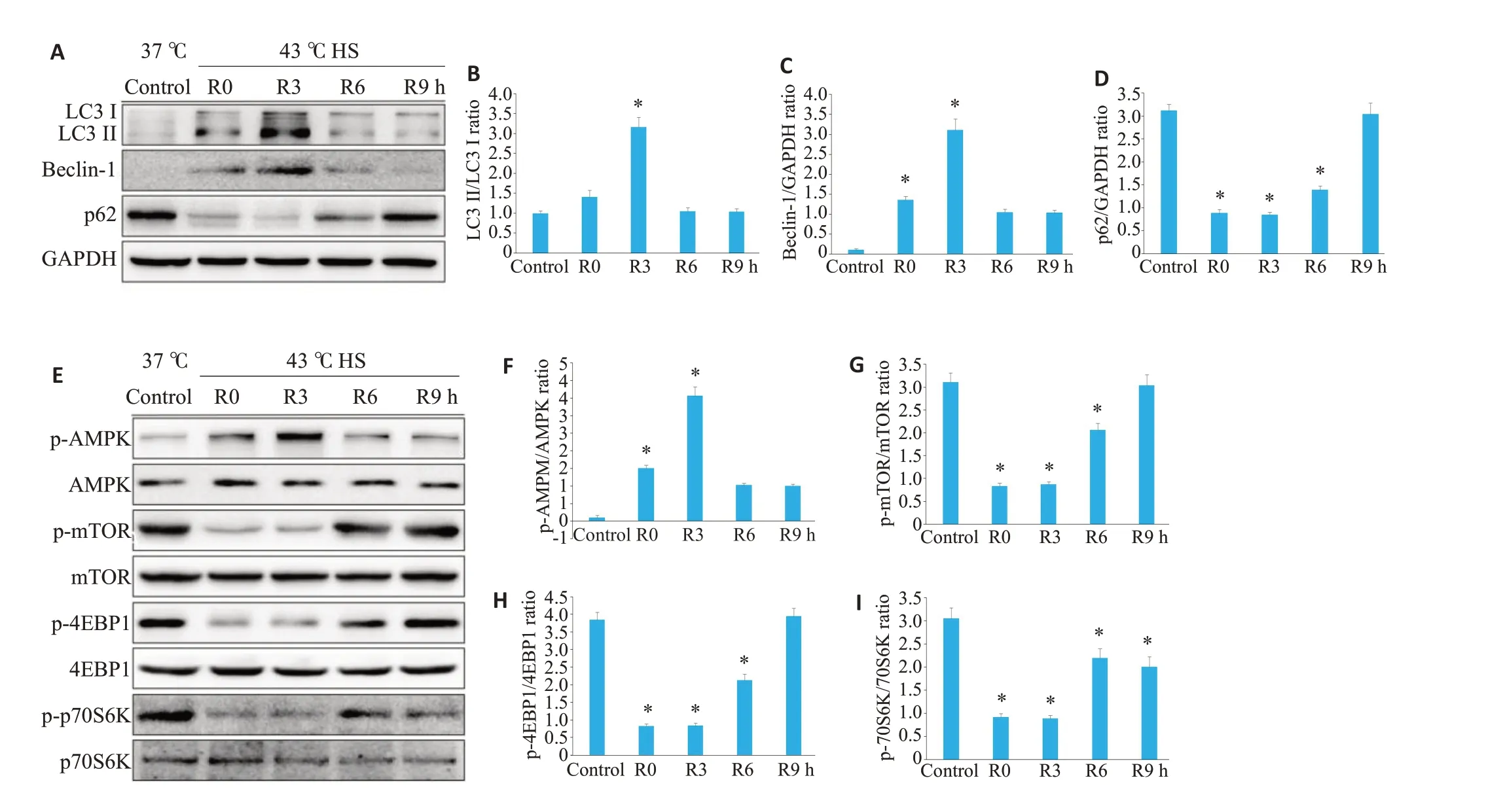
图2 热打击后MAECs中自噬相关蛋白LC3-Ⅱ、Beclin-1和P62以及AMPΚ/mTOR信号通路的变化情况Fig.2 Changes of autophagy-related proteins LC3-II,Beclin-1 and P62 and AMPK/mTOR signaling pathway in MAECs after HS.A:Levels of LC3-II,Beclin-1 and p62 detected with Western blotting.B:Quantification of the LC3-II and LC3-I ratio.C:Quantification of the Beclin-1 and GAPDH ratio.D:Quantification of p62 and GAPDH ratio.E:Levels of p-AMPK,AMPK,pmTOR,mTOR,p-4EBP1,4EBP1,p-p70S6K and p70S6K detected by Western blotting.F:Quantification of p-AMPK and AMPK ratio.G:Quantification of p-mTOR and mTOR ratio.H:Quantification of the p-4EBP1 and 4EBP1 ratio.I:Quantification of the p-70S6K and 70S6K ratio.*P<0.05 vs control group.
2.3 AMPΚ/mTOR信号通路介导了热打击诱导MAECs自噬
Western blot及confocal结果均显示,AMPΚ抑制剂CompoundC明显抑制了热打击诱导LC3-Ⅱ 蛋白的表达(图3A、B),而mTOR抑制剂rapamycin呈现相反作用,促进了热打击后LC3-Ⅱ蛋白的表达(图3C、D);Western blot结果显示,AMPΚ抑制剂Compound C明显抑制了热打击诱导Beclin-1蛋白的表达(图3 A、B),而mTOR抑制剂rapamycin促进了热打击后Beclin-1蛋白的表达(图3C、D),且与LC3-Ⅱ蛋白表达趋势呈一致性;而AMPΚ抑制剂Compound C明显促进了热打击诱导p62蛋白的表达(图3A、B),mTOR抑制剂rapamycin呈现相反作用(图3C、D),与LC3-Ⅱ和Beclin-1蛋白表达趋势相反。
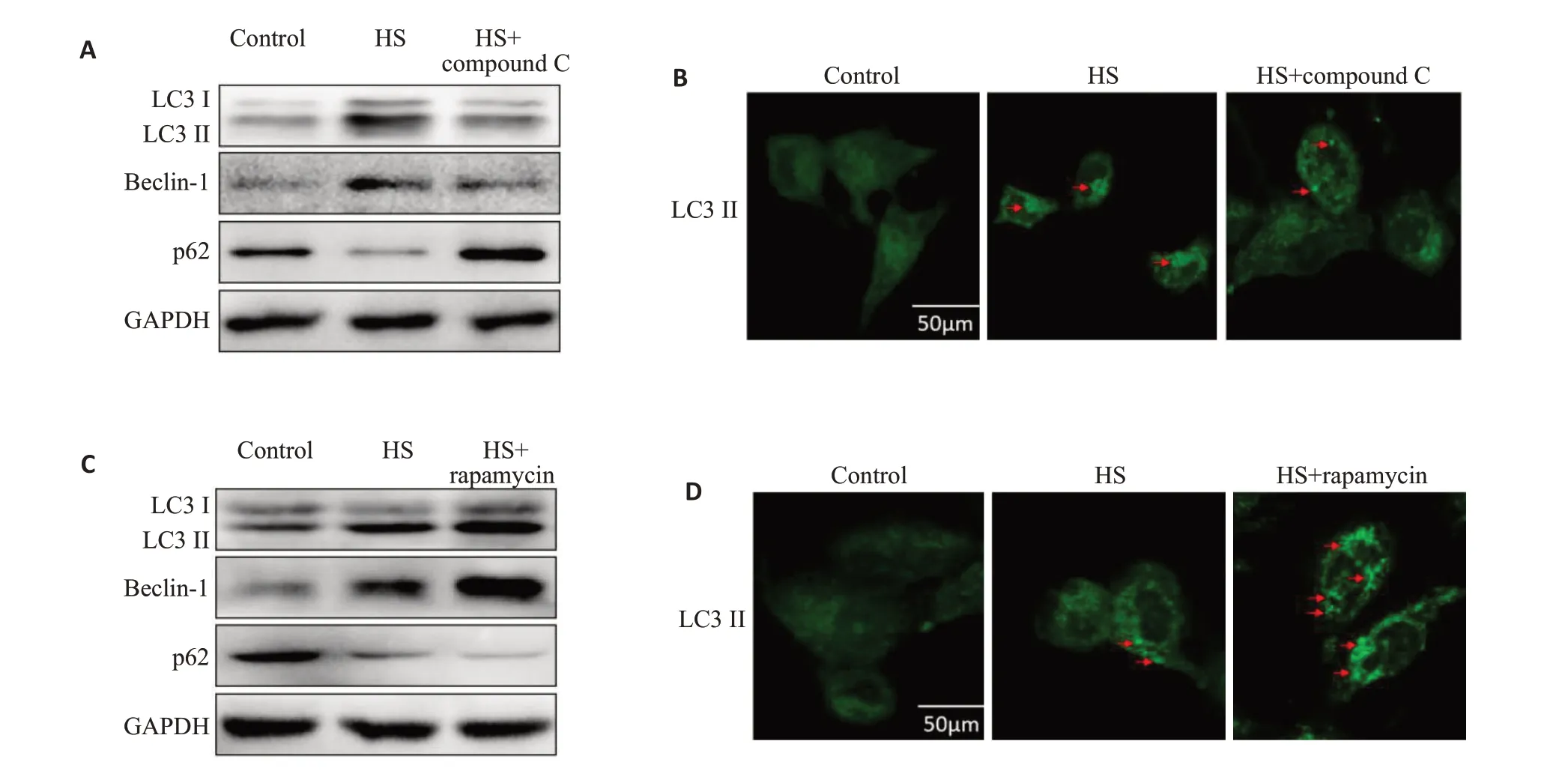
图3 AMPΚ/mTOR信号通路介导了热打击诱导MAECs自噬Fig.3 The AMPK/mTOR signaling pathway mediates HS-induced autophagy in MAECs.A:Effect of compound C on the expression of autophagy-related proteins LC3-II,Beclin-1 and P62 in MAECs detected by Western blotting;B:Effect of compound C on LC3-II expression in MAECs exposed to HS detected using confocal laser scanning microscopy.C:Effect of rapamycin on expressions LC3-II,Beclin-1 and P62 in MAECs detected by Western blotting.D:Effect of rapamycin on LC3-II expression in MAECs observed under confocal laser scanning microscope.
2.4 核外p53通过调控AMPΚ/mTOR信号通路介导自噬抑制,参与热打击诱导的MAECs和主动脉血管损伤
使用PFT后明显的促进了AMPΚ的磷酸化激活(图4A、B,P<0.05),抑制了mTOR以及其下游蛋白4EBP1和p70S6Κ的磷酸化(图4 A、C~E,P<0.05);使用PFT后明显的诱导了自噬的重要蛋白LC3-Ⅱ和Beclin-1的表达,并抑制了p62的表达(图4F~I,P<0.05);同时,使用PFT后明显提高了MAECs的细胞活力(图4J,P<0.05)。

图4 p53线粒体移位通过调控AMPΚ/mTOR信号通路介导的自噬参与热打击诱导的MAECs损伤Fig.4 p53 mitochondrial translocation is involved in HS-induced MAECs injury by regulating autophagy via the AMPK/mTOR signaling pathway.A:Levels of p-AMPK,AMPK,p-mTOR,mTOR,p-4EBP1,4EBP1,p-p70S6K and p70S6K detected by Western blotting.B:Quantification of the p-AMPK and AMPK ratio.C:Quantification of the p-mTOR and mTOR ratio.D:Quantification of the p-4EBP1 and 4EBP1 ratio.E:Quantification of the p-70S6K and 70S6K ratio.F:Levels of LC3-II,Beclin-1 and p62 detected by Western blotting.G:Quantification of the LC3-II and LC3-I ratio.H:Quantification of the Beclin-1 and GAPDH ratio.I:Quantification of the p62 and GAPDH ratio.J:Cell viability detected by CCK-8 assay.*P<0.05 vs control group.#P<0.05 vs with HS group.
进一步动物实验,分别使用AMPΚ 抑制剂Compound C、mTOR抑制剂(自噬激活剂)rapamycin、p53线粒体转位抑制剂PFT预处理小鼠,热打击后6 h分离小鼠主动脉血管发现,对照组小鼠主动脉血管内膜光滑,内膜下细胞排列整齐,内弹力层和中层平滑肌层次分明(图5A-a),未见明显凋亡细胞(图5B-a);热打击组小鼠主动脉血管染色不均,内皮细胞肿胀、脱落,内弹力膜断裂,平滑肌排列紊乱(图5A-b),可见明显的凋亡细胞(图B-b);热打击+AMPΚ抑制剂Compound C组小鼠主动脉血管损伤严重,内皮细胞明显脱落,内弹力膜断裂,平滑肌排列紊乱(图5A-c),可见大量凋亡细胞(图5B-c);与热打击组比较,热打击+mTOR 抑制剂(自噬激活剂)rapamycin 组和热打击+p53 线粒体转位抑制剂PFT组小鼠主动脉血管损伤有所减轻,内皮细胞肿胀、脱落有所改善(图5A-d、B-d),凋亡细胞减少(图5A-d、B-d)。
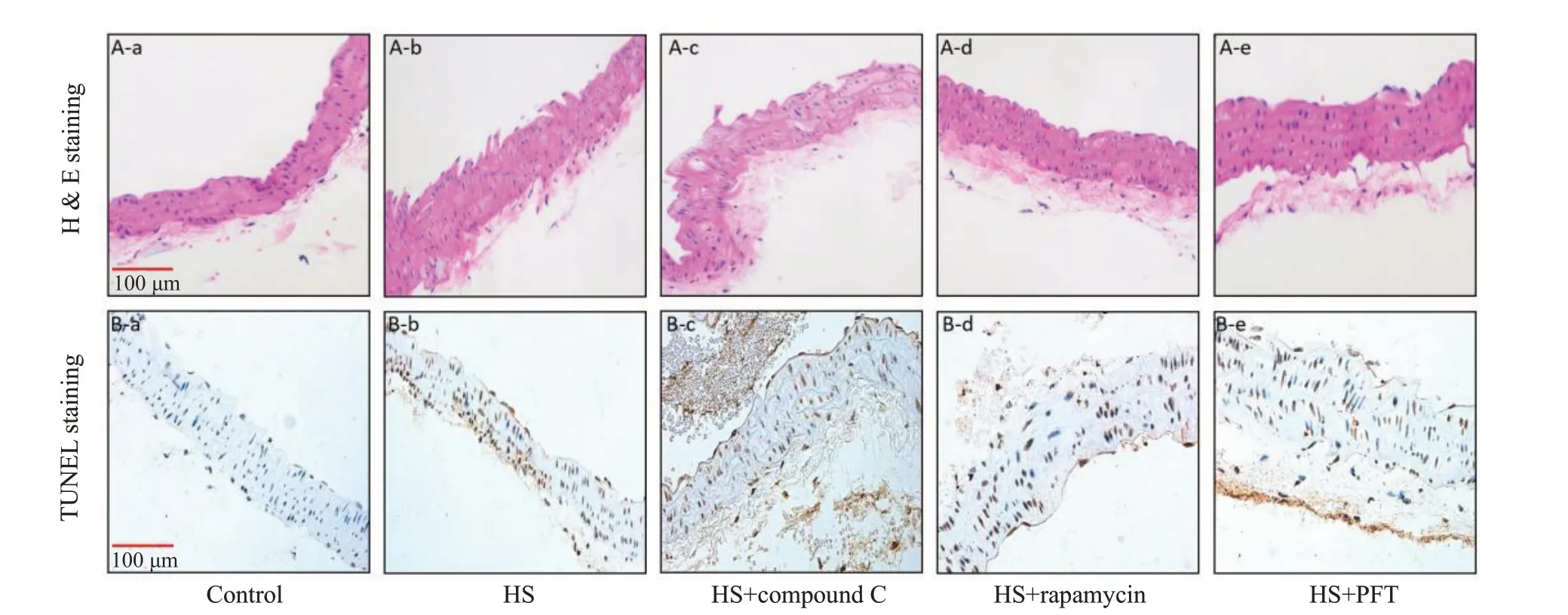
图5 p53线粒体移位通过调控AMPΚ/mTOR信号通路介导的自噬参与热打击诱导的小鼠主动脉血管损伤Fig.5 p53 mitochondrial translocation is involved in HS induced vascular injury in mouse aorta by regulating autophagy via the AMPK/mTOR signaling pathway.A:HE staining showing pathological changes of the aorta in each group;B:TUNEL staining for detecting aortic endothelial cell apoptosis in each group.
3 讨论
人体长时间在高温、高湿环境下可出现一系列不良应激反应,严重时诱发重症中暑的发生,并导致器官功能损害,直接威胁人们的生命[9,10]。在热损伤致中暑或重症中暑病理过程中,ⅤECs是最早出现形态和功能变化的细胞之一,其损伤可导致微循环障碍,启动弥漫性血管内凝血,参与全身炎症反应综合征,最终促使机体由热损伤进展为重症中暑甚至多器官功能障碍[10-13]。我们前期结果发现,在热打击的ⅤECs模型中,核外p53(p53线粒体移位)介导了细胞线粒体损伤、线粒体膜透化,最终诱导ⅤECs广泛损伤[1];深入的实验结果显示,核外p53主要是通过抑制自噬而导致了ⅤECs的广泛损伤[3],但是对于在这一过程中,关于核外p53介导自噬抑制的中间机制国内外尚无相关报道。在本试验中,我们通过分离原代的MAECs,发现热打击后细胞的活力降低;同时,通过分离主动脉内皮血管发现,热打击组小鼠主动脉血管染色不均,内皮细胞肿胀、脱落,内弹力膜断裂,平滑肌排列紊乱,并出现大量的凋亡细胞;进一步实验确认了热打击后可以诱导p53快速从胞质移位至线粒体[14],并随着复温时间的延长呈现逐渐增强效应,而热打击后MAECs的细胞活力出现复温时间依赖性降低。因而,热打击后可以导致明显的主动脉血管和细胞的损伤,并且与p53的线粒体移位有关。
自噬是普遍存在于真核细胞中一种进化上相当保守的代谢途径,其本质是一种细胞内的应激反应,在应激状态下可将细胞内变性、受损及非功能性的蛋白及亚细胞器等成分运送到溶酶体进行降解和循环利用,以维持细胞自身结构、功能和代谢的稳定[15-17]。正常的自噬过程对于维持细胞内环境的稳定性及细胞生命活动的顺利进行十分重要,但过度的自噬或自噬不足则会导致自噬应激,诱导细胞结构受损,继而引起细胞损伤[18,19]。过度的自噬可能诱导自噬性细胞死亡,因为过度的自噬活动会破坏大部分的胞质溶胶和细胞器,最终导致所有细胞功能完全崩溃。而细胞自噬不足时常引起损伤的细胞器及变性蛋白等不能被及时清除,内环境的稳定状态被破坏[20-22]。在本实验中,结果显示自噬重要蛋白LC3-Ⅱ和Beclin-1蛋白在热打击后开始表达,3 h达顶峰,6 h显著下降,而p62蛋白在热打击后被抑制,6 h后开始激活,与LC3-Ⅱ和Beclin-1蛋白表达呈现相反趋势。因此,热打击后早期即引起MAECs中自噬的激活,但随着复温时间的延长逐渐被抑制,推测热自噬抑制可能是导致打击后主动内皮血管和细胞损伤的重要机制。
AMPΚ是一种重要的能量感受器,当机体处于不利应激状态,AMPΚ被激活,AMPΚ活化后可同时关闭消耗ATP的合成代谢和开启ATP的分解代谢途径,通过这种机制维持细胞内代谢能量稳态[23]。AMPΚ在自噬过程中担当关键酶的角色,AMPΚ的激活可促使细胞自噬,降低组织细胞对能量的需求,同时也通过消化自身结构物质的机制来获取能量[24]。因此,AMPΚ的活化被认为在大多数的应激状态下对细胞具有保护作用。mTOR是与AMPΚ截然不同的能量感应器,在细胞自噬过程中具有门控作用,其活性是自噬体形成和成熟的关键,也是细胞自噬的负性调节因子[25]。mTOR是多数介导自噬信号通路的中间枢纽,因此mTOR被认为是细胞自噬最主要的抑制性通路。在AMPΚ介导的自噬信号通路中,mTOR 是其下游重要的信号分子。细胞遭受应激损伤过程中mTOR 受到抑制,其机制主要通过AMPΚ的激活[25]。在本实验中,结果显示热打击后即诱导了AMPΚ的磷酸化激活,3 h达高峰,6 h后被抑制;而mTOR以及其下游蛋白4EBP1和p70S6Κ的磷酸化在热打击后明显下调,6 h后表达开始上调,与AMPΚ的磷酸化呈现相反趋势。因此,热打击后早期激活了MAECs中AMPΚ/mTOR信号通路,但随着复温时间的延长逐渐被抑制,与热打击后MAECs 的自噬表达趋势呈现一致性。进一步实验发现,AMPΚ抑制剂Compound C明显抑制了热打击诱导的MAECs中LC3-Ⅱ和Beclin-1蛋白的表达,促进了热打击诱导p62蛋白的表达,并且使用Compound C预处理小鼠后,可导致热打击后小鼠主动脉血管损伤严重,内皮细胞明显脱落,内弹力膜断裂,平滑肌排列紊乱,出现大量凋亡细胞;而mTOR抑制剂rapamycin均呈现相反作用;因而,可以确认AMPΚ/mTOR 信号通路参与介导了热打击后主动脉血管和MAECs自噬的过程。
我们前期实验结果也发现核外p53可以通过抑制自噬诱导ⅤECs出现广泛损伤[3]。并且,p53通过磷酸化AMPΚ调控自噬抑制因子mTOR活性是其在核内发挥自噬促进功能的重要信号途径[5,26]。在细胞核内,p53可通过激活AMPΚ诱导SC1/TSC2复合体磷酸化激活,进而抑制mTOR,促进细胞自噬的发生[27]。此外,核内p53也能通过诱导氧化应激反应拮抗因子sestrins表达,并促使sestrins 与AMPΚ 结合,从而抑制mTOR 活性[28,29]。然而,目前对于核外p53抑制细胞自噬的分子机制尚不明确,相关也较少,也缺乏热损伤诱导核外p53抑制细胞自噬的相关研究。在本实验中,通过使用使用p53线粒体转位抑制剂PFT 预处理MAECs(已有研究证实PFT可有效消除核外p53对自噬的抑制作用,并且对核内p53 调控自噬作用无明显影响),PFT 明显促进AMPΚ的磷酸化激活,并抑制mTOR以及其下游蛋白4EBP1和p70S6Κ的磷酸化;使用PFT后也明显诱导了自噬的重要蛋白LC3-Ⅱ和Beclin-1的表达,并抑制了P62的表达;并且,使用PFT后明显提高了MAECs的细胞活力,减轻了小鼠主动脉血管损伤,内皮细胞肿胀、脱落有所改善,凋亡细胞减少。因而,核外p53主要通过抑制AMPΚ活性,激活mTOR信号,继而介导细胞自噬抑制,参与热打击诱导的主动脉血管和MAECs损伤。
本实验将热损伤后早期阶段作为研究切入点,并以ⅤECs 为研究的突破口,阐明了p53 主要通过调控AMPΚ/mTOR信号通路介导自噬抑制参与热打击诱导的MAECs损伤,但仍停留在实验现象的初步观察,仍需进一步深入的体内外实验完善目前结论。
