长牡蛎(Crassostrea gigas)黑色壳表面微生物多样性的研究*
2021-11-27李玲玲谢超伊宋宏策刘文娟黄宝玉张美溦刘雅琼王晓通
李玲玲 谢超伊 宋宏策 陈 熙 刘文娟 黄宝玉 张美溦 刘雅琼 魏 磊 王晓通
长牡蛎()黑色壳表面微生物多样性的研究*
李玲玲 谢超伊 宋宏策 陈 熙 刘文娟 黄宝玉 张美溦 刘雅琼 魏 磊①王晓通①
(鲁东大学农学院 烟台 264025)
为探究自然海区与室内养殖环境对软体动物壳色及壳表面细菌群落多样性的影响, 以黑色壳长牡蛎()为研究对象, 在自然海区与室内环境分别暂养30 d后, 对长牡蛎的壳色及壳表面细菌群落的多样性和差异细菌功能进行分析。结果表明, 室内养殖的黑壳长牡蛎出现较为明显的壳褪色现象, 其黑色素含量显著下降, 远低于自然海区养殖的黑壳长牡蛎。16S rRNA基因测序结果显示, 变形杆菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、蓝细菌(Cyanobacteria)为长牡蛎壳表面的主要优势菌, 其中变形杆菌的丰度超过50%。在门水平上, 放线菌(Actinobacteria)、俭菌超门(Parcubateria)和Bacterial rice cluster 1(BRC1)在室内、外养殖长牡蛎之间存在较为显著的差异(<0.05), 而具有降解作用的放线菌和俭菌超门细菌在室内养殖的黑壳长牡蛎壳表面丰度较高。在属水平上, 两组之间存在显著差异的细菌共计67种。该研究揭示了不同生境条件下, 黑壳长牡蛎壳表面的微生物群落物种组成及差异, 为解析长牡蛎壳色的形成机制提供了新的分析视角。
长牡蛎(); 细菌群落; 高通量测序; 黑色素; 壳色
颜色多态性是指在自然环境中, 一个物种的不同或同一群体中不同个体呈现出两种或两种以上可遗传的分离且不连续的颜色表型(Mckinnon, 2010)。贝壳颜色的不同会影响消费者的选择, 日常生活中, 消费者更倾向于购买颜色鲜艳的海产品(Brake, 2004)。研究发现, 贝壳颜色的不同对软体动物的生长、繁殖、抗氧化等多个方面都会产生影响(Adzigbli, 2020)。Ge等(2015)发现, 紫壳长牡蛎家系的存活率显著高于其他壳色。近年来, 随着软体动物选择育种研究的发展, 贝壳颜色作为一种重要的性状被广泛应用于多种软体动物的选育, 例如: 长牡蛎(), 虾夷扇贝()(Sun, 2015)、三角帆蚌()(Chen, 2017), 其中长牡蛎已被成功选育出黑壳、紫壳、橙壳、金壳和白壳等多种壳色的新品系(Zhu, 2018)。
软体动物的壳色通常是因不同色素沉积所形成, 例如: 吡咯、黑色素、卟啉、胆汁素、类胡萝卜素等, 其中黑色素的分布最为广泛(Stemmer, 2014; Williams, 2017; Liu, 2020)。黑色素可以保护生物免受寄生虫、污染物、低温、氧化应激和紫外线的影响, 参与器官的发育调控, 具有极为重要的作用(Dubey, 2014)。黑壳长牡蛎不仅外壳黑, 其外套膜及闭壳肌痕均呈现黑色, 富含黑色素, 具有较高的价值。研究表明, 高浓度双酚A (Bisphenol A, BPA)暴露后, 富含黑色素的黑壳长牡蛎组织未出现较为明显的病理变化, 表明黑壳长牡蛎与白壳长牡蛎相比可能具有更强的免疫力(姜秋云, 2019)。
微生物在动物的生存、生长代谢、营养物质吸收等过程中发挥着重要的作用(Bang, 2018)。随着高通量测序技术的发展, 越来越多的研究采用高通量测序的方法从种群结构、进化关系、群落多样性与环境之间的协作关系等多个方面对微生物进行研究。King等(2012)首次利用16S rRNA基因测序技术对美洲牡蛎()肠道和消化腺内的微生物种群组成进行探究。目前高通量测序技术在双壳贝类微生物多样性研究的应用主要集中在不同环境软体动物各组织微生物群落组成情况及差异细菌的作用分析(Madigan, 2014; Dubé, 2019; Horodesky, 2020); 致病菌对软体动物生长发育的影响等方面(Labare, 1990; Stevick, 2019)。对双壳贝类壳表面微生物的研究相对较少。已有的研究表明, 变形杆菌是双壳贝类壳表面的主要优势细菌, 美洲牡蛎壳表面微生物群落主要由α变形杆菌(Alphaproteobacteria)组成, 而加州贻贝()壳表面微生物则以γ变形杆菌(Gammaproteobacteria)为主(Pfister, 2014; Arfken, 2017)。
在黑壳长牡蛎的繁育过程中发现, 成体长牡蛎(壳长10—12 cm左右)经过约30 d室内养殖后, 其外壳表面颜色会变浅, 失去光泽, 出现壳褪色现象。在自然海区中养殖的黑壳长牡蛎则未发生外壳褪色的现象(图1)。一般而言, 不同的海区、不同的季节海洋水体理化性质具有较大的差异, 水体中微生物组成和丰度随季节等因素的变化有所改变(Wang, 2020)。受水环境的影响, 长牡蛎壳表面微生物组成上可能存在较大的差异。本研究以海区正常养殖及发生褪色现象的黑壳长牡蛎的壳表面微生物为研究对象, 对壳表面微生物的16S rRNA基因V3—V4区进行扩增并测序, 比较其微生物群落组成上的差异。从微生物多样性的角度, 探究了长牡蛎壳色的维持机制及褪色现象发生的原因。

图1 黑壳与褪色黑壳长牡蛎
1 材料与方法
1.1 实验材料
实验用黑壳长牡蛎均来自于山东省长岛县南长山岛连城湾海区养殖的个体选育家系。选取12只壳长(10—12 cm)、壳宽(4—6 cm)、壳高(2—3 cm)无显著差异的个体作为实验对象。随机挑选其中6只黑壳长牡蛎转入室内养殖实验室, 使用50 L玻璃缸养殖, 定时投喂密度为5.0×105个/mL的优质金藻, 喂食后3 h内换水一次, 养殖用海水取自山东省烟台市周边海区; 另外6只则继续在南长山岛海区中吊笼养殖。养殖期间, 自然海区及室内环境的盐度、温度及pH如表1所示。养殖30 d后, 分别对自然海区养殖未褪色黑壳长牡蛎样本(B组), 实验室内养殖褪色黑壳长牡蛎样本(F组)外壳表面微生物进行取样。取样时, 使用消毒棉签蘸取牡蛎壳表面的微生物, 随即将棉签保存至2 mL的冻存管中, 置于-80 °C低温冰箱保存。
表1 水环境基础指标

Tab.1 Basic indicators of water environment
1.2 壳色差异分析
利用Image-Pro Plus图像分析软件对实验所用黑壳长牡蛎外壳的光密度值(optical density values, ODV)进行测量, 用以表征壳表面的颜色变化。将长牡蛎外壳清洗干净后, 使用数码相机拍摄牡蛎壳照片。拍照时, 在相机的两边放置同等功率的日光灯, 以去除阴影的影响。测量/计数/尺寸/比例调节器设置为牡蛎壳的轮廓, 选定测定区域, 进行光密度的测定。强度校准设定为标准光密度, 灰度值转换为光密度值(ODV), 范围从0 (完全白色)到2.0 (完全黑色)。
1.3 DNA的抽提与PCR扩增测序
使用DNeasy®Power Water®Kit试剂盒(QIAGEN)进行基因组DNA的提取, 利用1%的琼脂凝胶电泳对基因组DNA的质量进行检测。16S rRNA V3-V4区的PCR扩增引物为338F(5′-ACTCCTACGGGAGGC AGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAA T-3′)。反应程序: 95 °C 30 min; 95 °C 30 s, 55 °C 30 s, 72 °C 45 s, 27个循环; 72 °C 10 min。PCR反应体系如表2所示。
表2 PCR反应体系

Tab.2 PCR reaction system
利用2%琼脂糖凝胶电泳检测PCR扩增产物的质量, 使用AxyPrep DNA胶回收试剂盒(Axygen Biosciences)切胶回收PCR产物, 最后使用Illumina MiSeq PE2500测序平台进行测序。
1.4 16S rRNA基因测序数据分析
为减少干扰数据的存在, 对原始测序数据进行如下的预处理: (1) 数据质控与优化统计, 使用Prinseq软件对测序数据质量进行控制, 去除序列长度小于50 bp的短片段和低复杂度序列。使用FLASH软件对测序数据进行拼接。(2) 去除嵌合体及靶区域外序列, 采用Mothur软件进行测序错误的矫正及去除嵌合体的操作。(3) 操作分类单元(operational taxonomic units, OTU)注释, 提取优化序列的非重复序列, 按照97%相似性对非重复序列(不含单序列)进行聚类。(4) 分类学注释, 利用Silva数据库(https:// www.arb-silva.de/版本号: 132)进行16S rRNA基因的比对。使用RDP classifier数据库(http://rdp.cme. msu.edu/misc/resources.jsp)进行物种分类学分析, 统计在各分类水平下微生物群落的组成情况。
预处理完成后, 进行如下的统计分析: (1) 使用Mothur软件对微生物群落进行Alpha多样性分析。使用Shannon指数绘制稀疏曲线, 以反映测序量的合理性。(2) 使用Qiime软件进行Beta多样性分析。(3) 利用Wilcox秩和检验(Wilcoxon rank-sum test)进行组间显著性差异检验, 使用FDR (false discovery rate)对值进行校正。(4) 利用线性判别分析(linear discriminant analysis effect size, LEfSe)筛选出在两组之间存在显著性差异, 具有生物学意义的物种, 并估算每个物种所产生的影响。比较策略选择all-against -all。(5) 基于皮尔逊相关系数分析环境因素与壳表面微生物群落之间的关系。
2 结果
2.1 壳色变化分析
ODV值的大小可间接反映黑壳表面的色素变化情况, 测定结果如图2所示。B组长牡蛎外壳表面的平均ODV值为0.68, F组长牡蛎外壳表面的平均ODV值为0.34, B组平均ODV值是F组的2倍, 两组样本平均ODV值存在极显著的差异(<0.01)。相比于自然海区养殖的长牡蛎, 室内养殖的长牡蛎外壳颜色变浅, 出现较为明显的褪色现象。

图2 灰度对比确定壳体色素沉积的平均光密度值(ODV)
注: **表示两组之间存在极显著差异(<0.01)
2.2 细菌群落Alpha多样性分析
测序结果显示, 有效序列数目在43 782—52 116之间, 序列平均长度为417.20 bp。本次测序共获得的4 728个OTU, 涵盖43个门, 99个纲, 199个目, 362个科、665个属, 1 271个种。两组样本的平均覆盖度(Coverage)均在98%以上, Coverage指数接近于1, 测序深度基本覆盖样本中的所有物种。Alpha多样性指数可反映微生物群落的丰富度与均匀度, 常使用Shannon指数、Simpson指数、ACE指数和Chao指数的大小表征。结果显示: B组的Shannon指数、ACE指数和Chao指数略高于F组, 而F组Simpson指数更高(表3)。Wilcoxon秩和检验显示, 两组样本的各Alpha多样性指数之间无显著性差异。稀疏曲线显示, 随着测序数据量的增加, 曲线逐渐趋于平缓, 当测序数据量在10 000时, 测序数据达到饱和(图3)。上述分析结果表明, 两组长牡蛎壳表面微生物群落物种组成较为丰富, 微生物群落的丰富度、均匀度均较高。
表3 细菌群落Alpha多样性指数

Tab.3 The alpha diversity index of bacterial communities

图3 16S rRNA基因测序稀疏曲线
2.3 Beta多样性分析
在OTU水平上, 基于Bray-Curtis距离算法, 利用非度量多维尺度分析(non-metric multidimensional scaling, NMDS)反映细菌群落的Beta多样性。NMDS分析结果显示, 组内各个样本点分布区域比较密集, 微生物群落结构相似, 差异较小(图4)。B组和F组的细菌形成了两个不同的群落空间结构, Beta多样性存在显著差异(Stress=0.124,<0.01)。使用相似性分析(analysis of similarities, ANOSIM), 探究两组样本间的微生物群落组成的差异性。ANOSIM分析结果表明, 两组样本的组间差异明显大于组内差异, 两组间的微生物群落结构差异显著(=0.307,<0.05)。上述分析结果显示, B组与F组之间的微生物群落分布差异较大, 而分组内的样本差异较小。
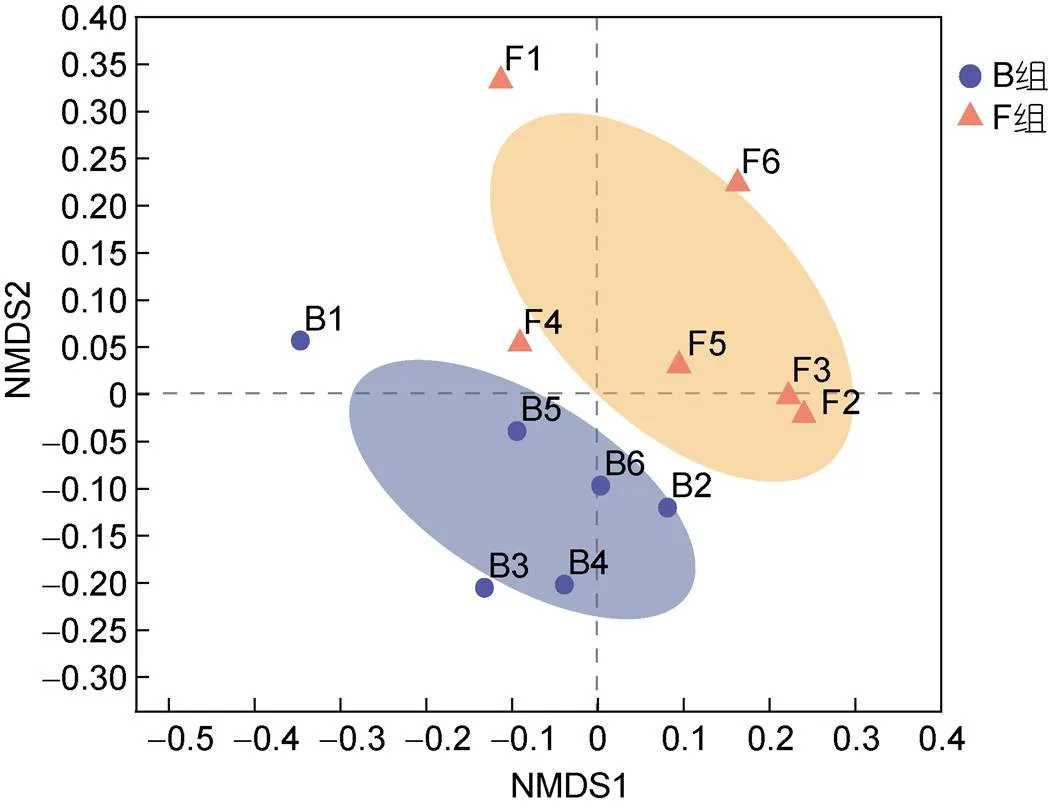
图4 非度量多维尺度分析(NMDS)
注: 图中每个点代表一个样品, 点与点之间的距离表示样品间物种组成差异程度, 同一颜色表示同一组的样品。紫色圆点为黑壳(B组), 橙色三角为褪色黑壳(F组)
2.4 微生物群落结构组成分析
Venn图可以直观的反映两组样本细菌群落OTU组成的差异性和重叠关系。两组样本微生物共有OTU数目为3 456 (图5)。在B组中, OTU的数目共计4 130, 独有OTU的数目674, 独有OTU占B组OTU总数的16.32%。独有OTU多隶属于酸杆菌门(Acidobacteria)、拟杆菌门(Bacteroidetes)、俭菌超门(Parcubateria)等。在F组中, OTU数目共计4 054, 独有OTU的数目为598, 独有OTU占F组OTU总数的14.75%。F组独有OTU多隶属于变形杆菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、Latescibacteria菌及浮霉菌门(Planctomycetes)等。从微生物的组成上看, 两组之间独有的OTU数目相对较少, 所占比例较小, 但分类较为丰富, 隶属于多个细菌门类。

图5 Venn图分析结果
对长牡蛎壳表面的微生物群落组成分析, 主要在门(phylum)和属(genus)两个分类学水平上展开。门水平上, 微生物群落主要由10个细菌门组成, 各样本中细菌丰度的差异如图6a所示。变形杆菌是主要的优势菌, 在两组样本中的丰度超过50.00%, 且以α变形杆菌的红杆菌科(Rhodobacteraceae)为主。拟杆菌门为第二优势菌, 在B组和F组中的丰度分别为19.70%和20.20%。其他优势菌如: 蓝细菌(Cyanobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)及浮霉菌门(Planctomycetes)等, 在样本中的丰度均超过1.00%, 但丰度较低(表4)。

图6 门水平(a)和属水平(b)上各样本细菌群落结构组成
表4 门水平优势菌的丰度

Tab.4 Abundance of phylum level dominant species
属水平上, 丰度排名为前25的细菌丰度差异结果如图6b所示。的丰度超过7.00%, 是主要的优势细菌。属水平上, 两组中丰度均超过1.00%的细菌有4种隶属于变形杆菌门, 它们分别是:、、和, 其中的丰度超过4.00%。细菌在两组样本中的丰度仅次于和(表5)。
表5 属水平优势菌的丰度
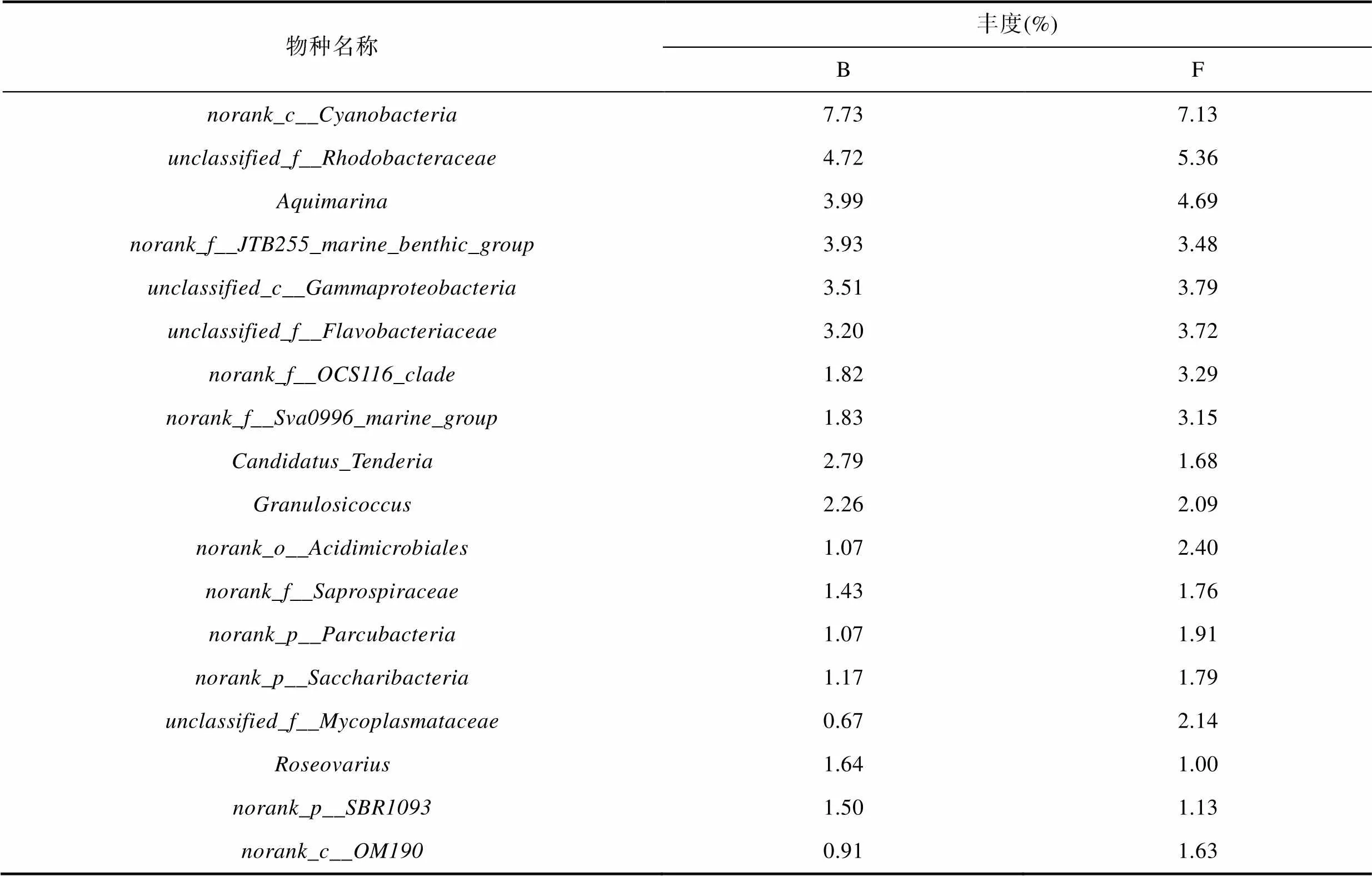
Tab.5 Abundance of dominant species at genus level
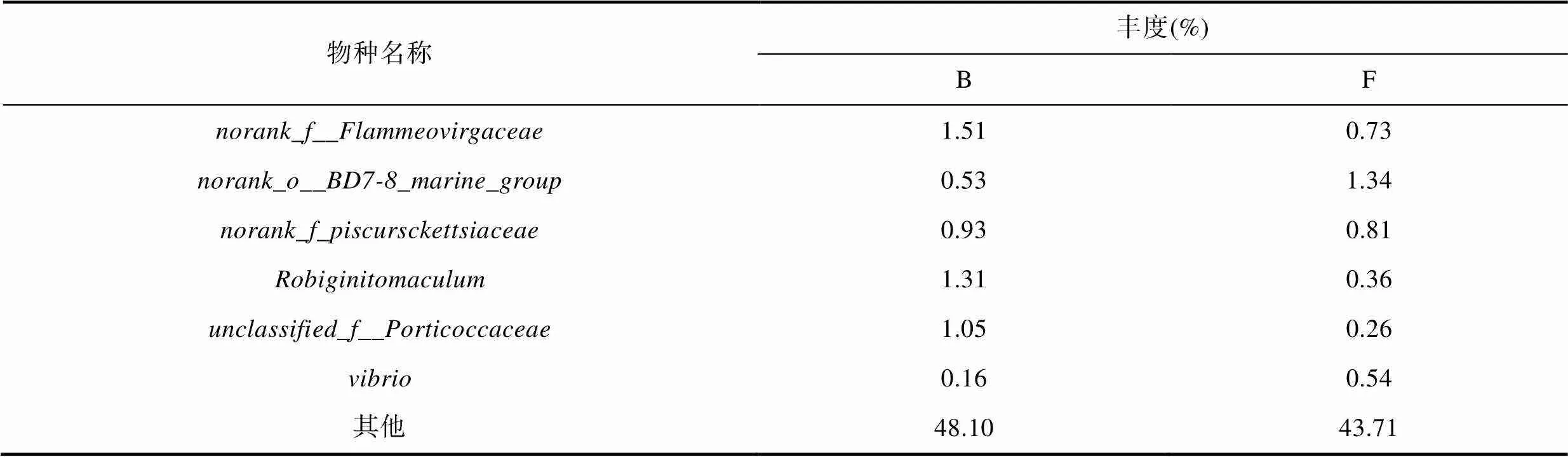
续表
2.5 组间差异显著性检验
Wilcox秩和检验结果显示: 放线菌门、俭菌超门和Bacterial rice cluster 1 (BRC1)在两组之间存在较为显著的差异(<0.05)。放线菌门和俭菌超门的细菌在F组的丰度极显著高于B组的丰度(<0.01), 且放线菌门和俭菌超门细菌的丰度之间存在极显著的相关性(< 0.01)。而BRC1菌则在B组的丰度(0.03%)显著高于F组(0.01%)(<0.05)。皮尔逊相关分析表明, 放线菌门、俭菌超门细菌的丰度与黑色素含量之间存在显著的负相关性(<0.05), BRC1细菌的丰度与黑色素的含量无明显的相关性。
属水平上, 67个属在两组间存在显著性差异(<0.05), 其中有19个属在两组间存在极显著性差异(<0.01)。如图7所示, 属水平上, 平均丰度处于前25的细菌, 有7种在两组之间存在显著差异, 其中、、、、在F组中丰度较高, 与B组存在较大差异。B组中, 玫瑰变色菌属()、的丰度较高, 与F组存在显著差异(<0.05)。除此之外, 科尔维尔氏菌属()在B组的丰度显著高于F组, 其相对丰度仅次于。、和细菌丰度与长牡蛎黑色素含量之间存在显著的负相关关系(<0.05)。其他几种差异细菌的丰度与长牡蛎黑色素含量之间无显著的相关性。在两组中, 存在显著差异的细菌属多隶属于变形杆菌门、放线菌门、浮霉菌门及拟杆菌门等, 变形杆菌门是丰度最高的细菌。在两组样本中丰度较高的物种, 比如:菌、和则在两组样本中不存在显著性差异。

图7 两组样本差异分析
注: **表示两组之间存在极显著差异(< 0.01), *表示两组之间存在显著差异(< 0.05)
LDA分布柱状图表明: 当LDA阈值设定为3.00时, 两组样本间共有17个物种的丰度存在显著差异(<0.05)(图8)。B组中、科尔维尔氏菌属、玫瑰变色菌属和等细菌分值较高, 其中的LDA分值最大(LDA=3.75), 产生的影响较大。F组的、、、等细菌的分值较高, 与B组存在显著差异(<0.05),是LDA分值最大的细菌(LDA=3.88)。
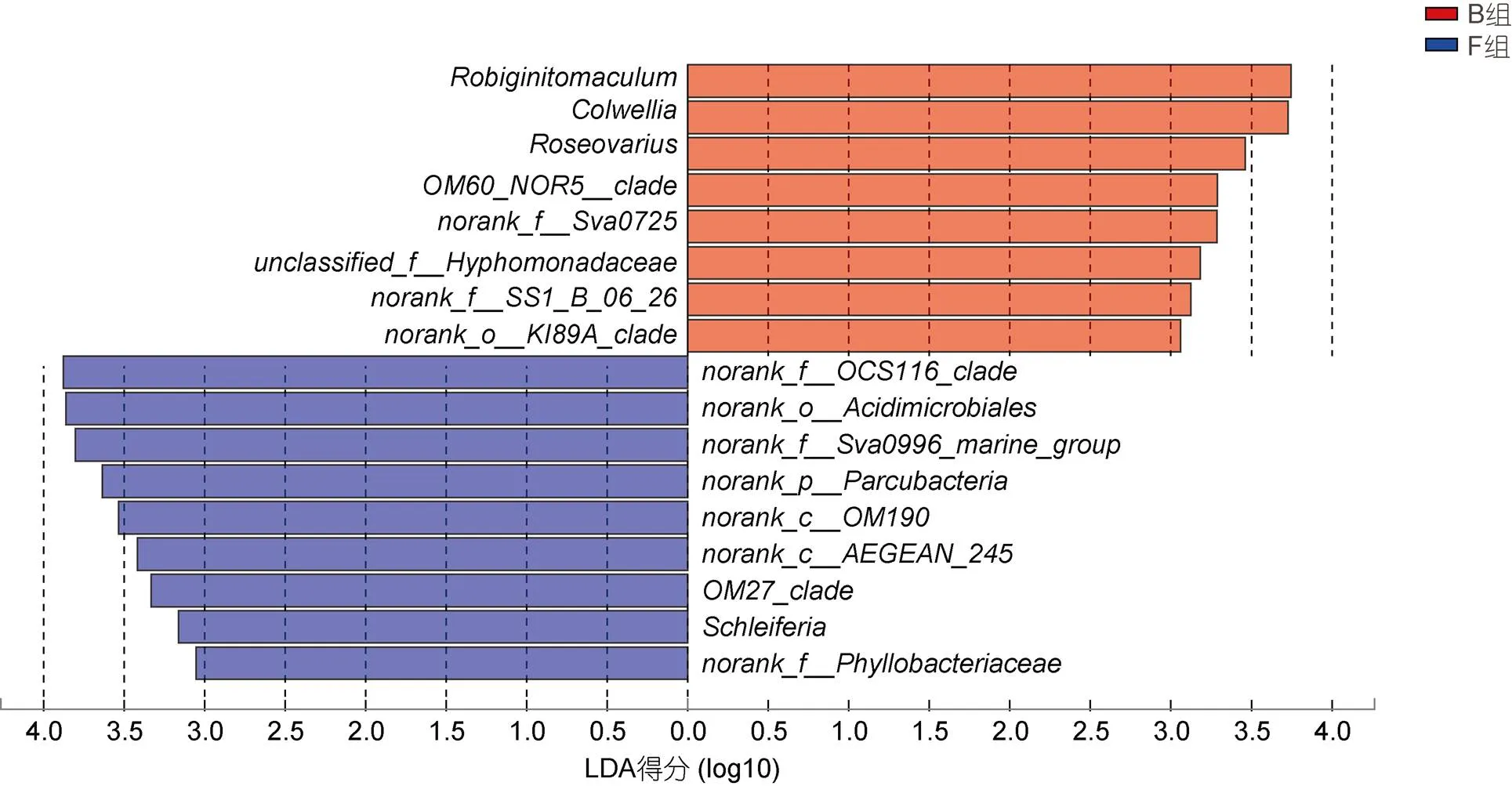
图8 LDA分布柱状图
2.6 环境因素对微生物群落的影响
利用皮尔逊相关系数反映微生物群落与水体环境之间的正负相关性。如图9所示: 属水平上, 相对丰度在前25的细菌中, 有5种细菌丰度与水体环境之间存在相关性。、、、、与盐度和温度之间存在显著的正相关, 与pH之间存在显著的负相关(<0.05)。而与pH之间存在显著的正相关, 与盐度和温度则呈现负相关关系(<0.05)。
3 讨论
研究表明, 外壳表面沉积不同种类的色素使贝类呈现出多彩的颜色。外壳表面黑色素的沉积使长牡蛎呈现黑色(Williams, 2017)。本实验所用长牡蛎富含黑色素, 不仅外壳黑, 外套膜和闭壳肌痕均呈现黑色。目前, 对于长牡蛎黑色素的发生机制尚未有明确的结论。Yu等(2017)的研究表明, 视黄醛脱氢酶、酪氨酸酶和细胞色素P450等基因家族可能在长牡蛎黑色素沉积过程中发挥重要作用。
已有对长牡蛎微生物群落的研究多集中在不同组织之间。在长牡蛎鳃、外套膜和血淋巴中, 变形杆菌为主要优势细菌; 而消化腺中变形杆菌、拟杆菌及放线菌的丰度较高(Fernández, 2014; Lokmer, 2016)。针对牡蛎壳表面微生物的研究相对较少。例如: Arfken等(2017)发现, 美国大西洋沿岸的美洲牡蛎()壳表面微生物群落组成主要以α变形杆菌中的鞘氨醇单胞菌科(Sphingomonadaceae)、Erythrobacteraceae科和红杆菌科(Rhodobacteraceae)为主。在韩国沿海废弃牡蛎壳表面, 变形杆菌所占比例达到了41%, 其次为拟杆菌(36.4%), 厚壁菌(Firmicutes)所占比例不足10%(Math, 2010)。本研究显示, 两种不同环境中的长牡蛎壳表面的主要优势菌同样为变形杆菌门, 其丰度超过50%, 以α变形杆菌为主。变形杆菌是海洋细菌中最重要的类群, α变形杆菌占海洋微生物总数的10%左右(Park, 2014; 张作峰等, 2020)。并且山东烟台周边海域海洋水体中α变形杆菌亦是第一优势菌(张艳, 2010)。因此, 长牡蛎所处水体环境与壳表面优势细菌的组成具有极大的相似性。
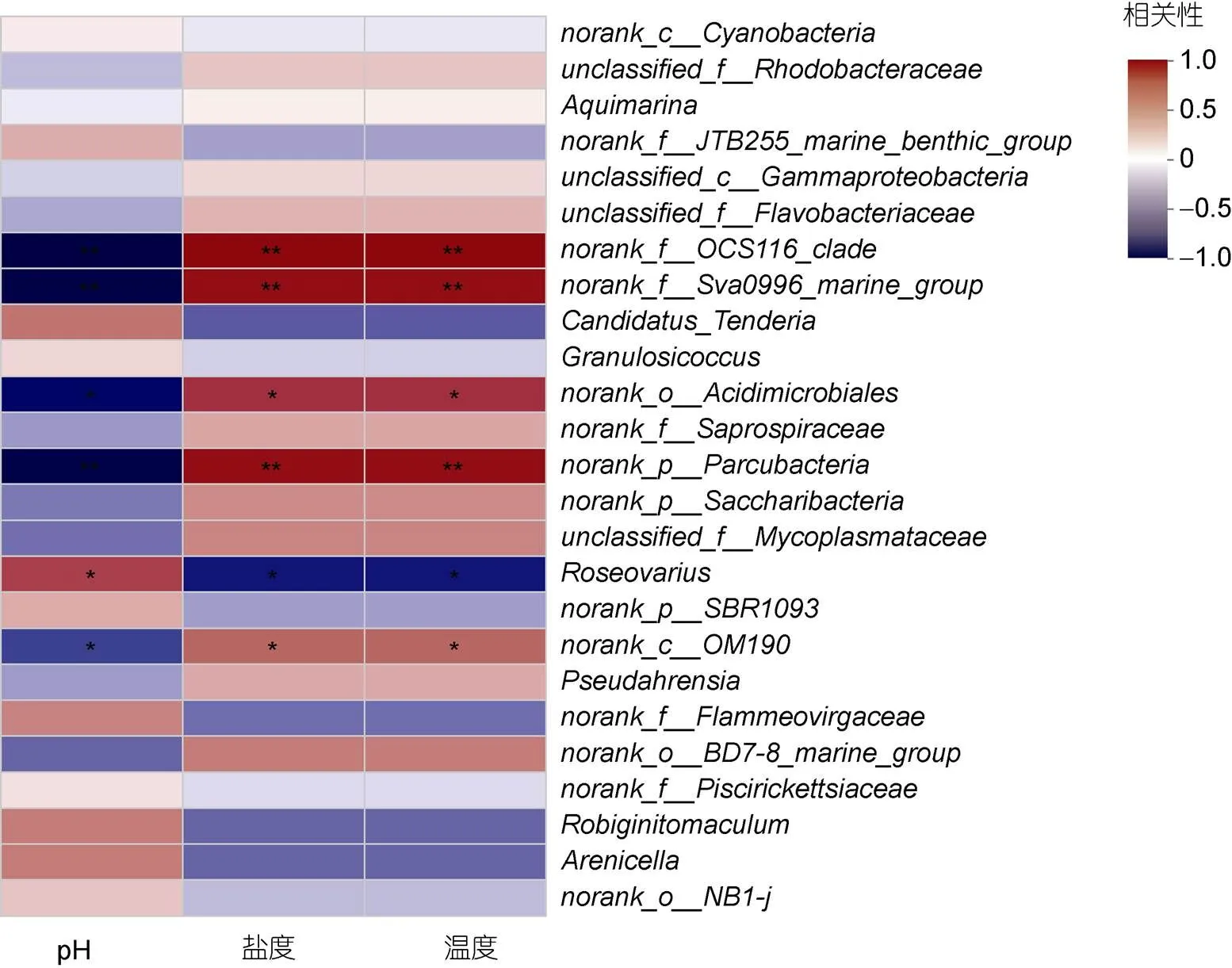
图9 水体性质与微生物群落之间的相关性
注: **表示两组之间存在极显著差异(< 0.01), *表示两组之间存在显著差异表示(< 0.05)
进一步的分析显示, 在微生物群落的组成上, 两种水体环境中长牡蛎壳表面的微生物群落组成上存在一定差异。门水平上, BRC1细菌在B组的丰度远高于F组, 而放线菌门和俭菌超门在F组的丰度更高。
BRC1细菌广泛存在于富含有机物的环境中(Kadnikov, 2019), 目前尚未成功培养出BRC1细菌门的菌株, BRC1细菌的具体功能尚不清晰, 仍需进一步的研究。在F组中, 放线菌所占比例达到了6.14%, 约是B组的两倍。放线菌广泛存在于各种环境中, 具有强大的生物合成潜力, 能够产生具有较强抗菌活性的次级代谢产物。放线菌还具有降解有机物的作用, 其产生的水解酶可降解各种生物聚合物, 例如: 纤维素、木聚糖和几丁质, 在氮碳循环过程中发挥着重要的作用(Manivasagan, 2013; Fernández, 2014)。除放线菌外, 在F组的长牡蛎壳表面发现, 俭菌超门的丰度(2.19%)高于B组(1.20%)。俭菌超门细菌的基因组几乎都在1 Mb以下, 缺乏能量代谢的关键基因。因此, 俭菌超门细菌无法单独生活, 需要与其他微生物共生生活, 例如放线菌等(Nelson, 2015)。相关性分析结果表明, 俭菌超门与放线菌的丰度存在极显著的相关性(<0.01)。俭菌超门的部分细菌同样具有降解功能, 可以降解环境中的纤维素和甲壳素(Nelson, 2015; 陶晔等, 2020)。长牡蛎壳中的黑色素不溶于水, 与蛋白质、多糖等生物聚合物结合紧密(何成等, 2017)。F组中, 放线菌和俭菌超门细菌的大量聚集可能促进了长牡蛎壳表面生物聚合物的降解过程, 从而发生了壳褪色现象。
门水平上, 存在显著差异的细菌的数目较少, 多数细菌的丰度无较大的差异。属水平上, 差异细菌的数目较多, 超过50个属在两组之间存在显著的差异。相比于F组, 玫瑰杆菌属、科尔维尔氏菌属和在B组长牡蛎黑壳表面的丰度均较高。
玫瑰杆菌属广泛存在于各种海洋介质中, 比如海水、沉积物、海冰等。玫瑰杆菌属通过脱甲基途径和裂解途径将海洋中的有机硫化物二甲基疏基丙酸内盐(Dimethylsulfoniopropionate, DMSP)代谢分解, 参与硫循环(Sun, 2016)。在本次测序中, 玫瑰杆菌属在B组的丰度为1.64%, 高于F组的1.00%。推测其可能的原因是水体环境的不同造成细菌丰度的差异。科尔维尔氏菌属是嗜冷菌, 在海冰、极地沉积物、深海海沟中均有发现(Nogi, 2004)。科尔维尔氏菌属具有降解碳氢化合物的作用, 在溢油环境中, 可大量检出(Gutierrez, 2013; Mason, 2014)与科尔维尔氏菌的作用相似, 同样具有降解碳氢化合物的作用。富含碳氢化合物的沉积物中菌的丰度和多样性都会升高(Guibert, 2012; Hestetun, 2015)。
在南长山岛养殖的黑壳长牡蛎壳表面, 科尔维尔氏菌和的丰度均高于室内养殖的长牡蛎。可能是由于外源水体的排放, 导致渤海湾水体中的碳氢化合物含量较高, 促进了具有降解功能细菌的繁殖, 例如科尔维尔氏菌和。因此, 水体环境可能对细菌的丰度产生较大的影响。然而, 由于科尔维尔氏菌属和属细菌难以独立培养, 在长牡蛎壳色维持方面所起到的作用尚不明确, 仍需进一步的探索。
微生物群落的组成是动态的, 受地理环境的影响较大, 随盐度、温度、pH值等环境因素的变化而变化(Prieur, 1990; Harris, 1993; Jing, 2019)。自然海区养殖的长牡蛎生活环境复杂多变, 在潮汐、波浪等风力或水力的作用下, 水体不断混合, 水体微生物处于不断的变化之中。皮尔逊相关性分析结果表明, 在两组样本中存在显著差异细菌的丰度与水体环境之间存在较强的相关性。其中、、、、细菌的丰度与盐度和温度之间存在显著的正相关关系, 与pH之间存在显著的负相关关系(<0.05)。而细菌的丰度与pH之间存在显著的正相关关系, 与盐度和温度则呈现显著的负相关关系(< 0.05)。因此, 环境条件与微生物丰度之间相关性较强, 并可影响微生物的丰度。
4 结论
与自然海区相比, 室内养殖的长牡蛎发生了壳褪色现象, 且黑色素含量显著下降。壳表面微生物多样性分析表明, 两种环境中养殖的长牡蛎微生物群落在种类组成上无较大的差异, 但是细菌的丰度有所不同。变形杆菌门是长牡蛎壳表面微生物群落的主要优势菌, 其丰度在两组中超过50%。褪色黑壳长牡蛎表面具有降解作用的细菌丰度较高, 例如放线菌门和俭菌超门, 推测其可能参与长牡蛎壳褪色过程, 环境条件的不同可影响长牡蛎壳表面细菌的丰度。本研究从微生物多样性的角度, 解析了黑壳长牡蛎壳表面褪色现象的可能原因。
何 成, 于文超, 蔡忠强等, 2017. 酸碱粗提配合超声破碎法制取可溶性长牡蛎()黑色素的研究. 海洋与湖沼, 48(3): 634—639
张 艳, 2010. 山东近岸海域水体细菌多样性研究. 青岛: 中国海洋大学硕士学位论文
张作峰, 曾令宇, 王兆守等, 2020. 功能化石墨烯对海洋环境中细菌群落结构及生长的影响. 厦门大学学报(自然科学版), 59(2): 271—277
姜秋云, 2019. 黑壳与白壳长牡蛎免疫力的比较分析. 上海: 上海海洋大学硕士学位论文
陶 晔, 邢 鹏, 2020. 候选门级辐射类群(CPR)细菌研究进展. 微生物学报, 60(6): 1284—1303
葛建龙, 李琪, 于红等, 2015.长牡蛎3种壳色家系间杂交子代生长和存活比较.水产学报, 39(3):345—352
Adzigbli L, Wang Z M, Li J H, 2020. Survival, retention rate and immunity of the black shell colored stocks of pearl oysterafter grafting operation. Fish & Shellfish Immunology, 98: 691—698
Arfken A, Song B, Bowman J S, 2017. Denitrification potential of the eastern oyster microbiome using a 16S rRNA gene based metabolic inference approach. PLoS One, 12(9): e0185071
Bang C, Dagan T, Deines P, 2018. Metaorganisms in extreme environments: do microbes play a role in organismal adaptation?. Zoology, 127: 1—19
Brake J, Evans F, Langdon C, 2004. Evidence for genetic control of pigmentation of shell and mantle edge in selected families of Pacific oysters,. Aquaculture, 229(1—4): 89—98
Chen X J, Liu X J, Bai Z Y, 2017.andof, novel tyrosinase and tyrosinase-related protein genes involved in nacre color formation. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 204: 1—8
Dubé C E, Ky C L, Planes S, 2019. Microbiome of the black-lipped pearl oyster, a multi-tissue description with functional profiling. Frontiers in Microbiology, 10: 1548
Dubey S, Roulin A, 2014. Evolutionary and biomedical consequences of internal melanins. Pigment Cell & Melanoma Research, 27(3): 327—338
Fernández N T, Mazón-Suástegui J M, Vázquez-Juárez R, 2014. Changes in the composition and diversity of the bacterial microbiota associated with oysters (,and) during commercial production. FEMS Microbiology Ecology, 88(1): 69—83
Guibert L M, Loviso C L, Marcos M S, 2012. Alkane biodegradation genes from chronically polluted subantarctic coastal sediments and their shifts in response to oil exposure. Microbial Ecology, 64(3): 605—616
Gutierrez T, Singleton D R, Berry D, 2013. Hydrocarbon-degrading bacteria enriched by theoil spill identified by cultivation and DNA-SIP. The ISME Journal, 7(11): 2091—2104
Harris J M, 1993. The presence, nature, and role of gut microflora in aquatic invertebrates: a synthesis. Microbial Ecology, 25(3): 195—231
Hestetun J T, Fourt M, Vacelet J, 2015. Cladorhizidae (Porifera, Demospongiae, Poecilosclerida) of the deep Atlantic collected during Ifremer cruises, with a biogeographic overview of the Atlantic species. Journal of the Marine Biological Association of the United Kingdom, 95(7): 1311—1342
Horodesky A, Castilho-Westphal G G, Pont G D, 2020. Metagenomic analysis of the bacterial microbiota associated with cultured oysters (sp.) in estuarine environments. Anais da Academia Brasileira de Ciencias, 92(S1): e20180432
Jing X Y, Gou H L, Gong Y H, 2019. Seasonal dynamics of the coastal bacterioplankton at intensive fish-farming areas of the Yellow Sea, China revealed by high-throughput sequencing. Marine Pollution Bulletin, 139: 366—375
Kadnikov V V, Mardanov A V, Beletsky A V, 2019. Phylogeny and physiology of candidate phylum BRC1 inferred from the first complete metagenome-assembled genome obtained from deep subsurface aquifer. Systematic and Applied Microbiology, 42(1): 67—76
King G M, Judd C, Kuske C R, 2012. Analysis of stomach and gut microbiomes of the eastern oyster () from coastal Louisiana, USA. PLoS One, 7(12): e51475
Labare M P, Weiner R M, 1990. Interactions between, oyster larvae, and hydrophobic organophosphate pesticides. Applied and Environmental Microbiology, 56(12): 3817—3821
Liu H X, Tan K S, Zhang X X, 2020. Comparison of gut microbiota between golden and brown noble scallopand its association with carotenoids. Frontiers in Microbiology, 11: 36
Lokmer A, Kuenzel S, Baines J F, 2016. The role of tissue-specific microbiota in initial establishment success of Pacific oysters. Environmental Microbiology, 18(3): 970—987
Madigan T L, Bott N J, Torok V A, 2014. A microbial spoilage profile of half shell Pacific oysters () and Sydney rock oysters (). Food Microbiology, 38: 219—227
Manivasagan P, Venkatesan J, Sivakumar K, 2013. RETRACTED: marine actinobacterial metabolites: current status and future perspectives. Microbiological Research, 168(6): 311—332
Mason O U, Han J, Woyke T, 2014. Single-cell genomics reveals features of aspecies that was dominant during the Deepwater Horizon oil spill. Frontiers in Microbiology, 5: 332
Math R K, Islam A, Hong S J, 2010. Metagenomic characterization of oyster shell dump reveals predominance ofbacteria. Microbiology, 79(4): 509—519
Mckinnon J S, Pierotti M R, 2010. Colour polymorphism and correlated characters: genetic mechanisms and evolution. Molecular Ecology, 19(23): 5101—5125
Nelson W C, Stegen J C, 2015. The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Frontiers in Microbiology, 6: 713
Nogi Y, Hosoya S, Kato C, 2004.sp. nov., a novel piezophilic species from deep-sea sediments of the Japan Trench. International Journal of Systematic and Evolutionary Microbiology, 54(5): 1627—1631
Park S, Jung Y T, Won S M, 2014.sp. nov., a member of the familyisolated from a brown algae reservoir and emended description of the genus. Antonie van Leeuwenhoek, 106(5): 845—852
Pfister C A, Gilbert J A, Gibbons S M, 2014. The role of macrobiota in structuring microbial communities along rocky shores. PeerJ, 2: e631
Prieur D, Mével G, Nicolas J L, 1990. Interactions between bivalve molluscs and bacteria in the marine environment. Oceanography and Marine Biology: an Annual Review, 28: 277—352
Stemmer K, Nehrke G, 2014. The distribution of polyenes in the shell offrom North Atlantic localities: a confocal Raman microscopy study. Journal of Molluscan Studies, 80(4): 365—370
Stevick R J, Sohn S, Modak T H, 2019. Bacterial community dynamics in an oyster hatchery in response to probiotic treatment. Frontiers in Microbiology, 10: 1060
Sun J, Todd J D, Thrash J C, 2016. The abundant marine bacteriumsimultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethio. Nature Microbiology, 1(8): 16065
Sun X J, Yang A G, Wu B, 2015. Characterization of the mantle transcriptome of Yesso Scallop (): identification of genes potentially involved in biomineralization and pigmentation. PLoS One, 10(4): e0122967
Wang Y P, Li G H, Shi F, 2020. Taxonomic diversity of pico-/nanoeukaryotes is related to dissolved oxygen and productivity, but functional composition is shaped by limiting nutrients in eutrophic coastal oceans. Frontiers in Microbiology, 11: 601037
Williams S T, 2017. Molluscan shell colour. Biological Reviews, 92(2): 1039—1058
Yu W C, He C, Cai Z Q, 2017. A preliminary study on the pattern, the physiological bases and the molecular mechanism of the adductor muscle scar pigmentation in Pacific oyster. Frontiers in Physiology, 8: 699
Zhu Y J, Li Q, Yu H, 2018. Biochemical composition and nutritional value of different shell color strains of Pacific Oyster. Journal of Ocean University of China, 17(4): 897—904
MICROBIAL DIVERSITY ON SURFACE OF BLACK-SHELL
LI Ling-Ling, XIE Chao-Yi, SONG Hong-Ce, CHEN Xi, LIU Wen-Juan, HUANG Bao-Yu, ZHANG Mei-Wei, LIU Ya-Qiong, WEI Lei, WANG Xiao-Tong
(School of Agriculture, Ludong University, Yantai 264025, China)
To explore the effects of natural water and indoor environment on the shell color and bacterial community diversity of molluscs, the melanin-richwas cultured in the natural water and indoor environment for 30 days. The diversity of bacterial community structure and the function of different bacteria on the shell color and shell surface of thewas analyzed. Results show that the black-shelledcultured indoor showed obvious shell fading phenomenon and the melanin content was significantly decreased, which was much lower than the black-shelledcultured in natural water. 16S rRNA gene sequencing showed that Proteobacteria, Bacteroidetes, and Cyanobacteria were the main dominant bacteria on the surface ofshells and the abundance of Proteobacteria exceeded 50%. At the phylum level, Actinobacteria, Parcubateria, and Bacterial rice cluster 1 (BRC1) were significantly different between the two groups (< 0.05). Among them, the Actinobacteria and Parcubateria bacteria with degrading effect was relatively high in the surface abundance of the black-shelledgrown indoor. At the genus level, there were 67 species of bacteria with significant differences between the two groups. This study revealed the species composition and differences of the microbial community on surface of oyster shell under different habitat conditions, and provided a new analytical perspective for the oyster shell color manipulation.
; bacterial community; high-throughput sequencing; melanin; shell color
* 国家自然科学基金项目, 41906088号; 国家自然科学基金项目, 41876193号; 山东省高等学校“青创科技计划”, 2019KJF004号; 山东省贝类产业技术体系, SDAIT-14号。李玲玲, 硕士研究生, E-mail: lilingling0603@126.com; 并列第一作者: 谢超伊, 硕士研究生, E-mail: 2433040312@qq.com
魏 磊, 副教授, E-mail: lei819@126.com; 王晓通, 教授, E-mail: wangxiaotong999@163.com
2021-04-06,
2021-06-08
S917
10.11693/hyhz20210400082