Microscopic Mechanism on Giant Photoeffect in Proton Transport Through Graphene Membranes
2021-11-22LimingGuanBeidouGuoXinruiJiaGuancaiXieJianRuGong
Liming Guan , Beidou Guo , Xinrui Jia , Guancai Xie , Jian Ru Gong,*
1 Chinese Academy of Sciences (CAS)Center for Excellence in Nanoscience, CAS Key Laboratory of Nanosystem and Hierarchy Fabrication, National Center for Nanoscience and Technology, Beijing 100190, China.
2 University of Chinese Academy of Sciences, Beijing 100049, China.
Abstract:Graphene monolayers are permeable to thermal protons and impermeable to other atoms and molecules, exhibiting their potential applications in fuel cell technologies and hydrogen isotope separation.Furthermore, the giant photoeffect in proton transport through catalytically activated graphene membranes was reported by Geim et al. Their experiment showed that the synergy between illumination and the catalytically active metal plays a key role in this photoeffect. Geim et al.suggested that the local photovoltage created between metal nanoparticles and graphene could funnel protons and electrons toward the metal nanoparticles for the production of hydrogen, while repelling holes away from them, causing the giant photoeffect. However, based on static electric field theory, this explanation is not convincing and the work lacks an analysis on the microscopic mechanism of this effect. Herein, we provide the exact microscopic mechanism behind this phenomenon. In semi-metal pristine graphene, most photon excited hot electrons relax to lower energy states within a timescale of 10-12 s, while the typical timescale of a chemical reaction is 10-6 s. Thus, hot electrons excited by incident photons relax to lower energy states before reacting with protons through the graphene. When graphene is decorated with metal, electron transfer between the graphene and the metal, induced by different work functions, would result in the formation of interface dipoles. When using metals such as Pt, Pd, Ni, etc., which can strongly interact with graphene, local dipoles form. Protons are trapped around the negative poles of the local dipoles, while electrons are around the positive poles. Upon illumination, the electrons are excited to metastable excited states with higher energy levels. Due to the energy barriers around them, the free electrons in the metastable excited states will have a relatively longer lifetime, which facilitates the production of hydrogen through their effective reaction with protons that permeated through the graphene.The concentration of high-energy electrons under illumination was estimated, and the results showed that more electrons are energized to the excited state with strong illumination. According to the analysis, the giant photoeffect in proton transport through the catalytically activated graphene membrane is attributed to long-lived hot electrons and a fast proton transport rate. Since there is no change in the activation energy of the reaction, the metal catalyst increases the rate of the reaction by increasing the number of successful collisions between the reactants to produce the significant photoeffect.This might lead to a new microscopic mechanism that clarifies the role of the catalyst in improving the efficiency of photo(electro)catalytic reactions.
Key Words:Graphene;Proton transport;Dipole;Hot electron;Hydrogen
1 Introduction
Graphene monolayers have been shown to be permeable to thermal protons, while impermeable to other atoms and molecules, displaying their potential applications such as fuel cell technologies and hydrogen isotope separation1–4. In theory,first-principle calculations have shown that the energy barrier for proton permeation is in the range of 1.17–3.0 eV depending on the adsorption states of proton5–7. As for the experiment, it was found that proton permeation through graphene can be thermally activated with an energy barrier of 0.8 eV2. Further measurements using the hydrogen isotope deuterium demonstrated that this barrier is actually ~1 eV due to the increased energy of incoming protons by quantum oscillation3.These discrepancies between the barriers found in theory and those in experiments triggered debates about the exact microscopic mechanism behind the proton permeation through graphene. Various mechanisms, such as hydrogenation of graphene, quantum tunneling of thermal protons, have been used to explain such proton permeation8–10. Furthermore, Geimet al.11reported the giant photoeffect in proton transport through catalytically-activated graphene. In their experiment, protons transport from a proton source of the PdHxelectrode through a monolayer graphene under a bias voltage, then react with electrons on the other side of graphene to generate hydrogen when catalytically-active metal nanoparticles were decorated on graphene in the dark. Under illumination, a strongly enhanced proton current as well as hydrogen flux can be observed, and the Faradaic efficiency is 100% at all proton current densities and the activation energy does not change noticeably. In contrast, if graphene is not effectively catalytically-activated, there is no enhancement of the proton current under illumination.Obviously, a synergy of illumination and the catalytic-active metal plays a key role in this photoeffect.
On the basis of the facts in the experiment, a few possible mechanisms were first ruled out for explaining the photoeffect11.They include (1)plasmonic effect, due to the lack of response in the presence of plasmon-active Au nanoparticles12,13, (2)photoinduced electrolysis of water, since the applied voltage is well below the thermodynamic voltage for water splitting and graphene is impermeable to hydrogen11,14,15, and (3)photoinduced hydrogenation of graphene, because the expected reduction in the energy barrier, that is, the activation energy, is not observed in the experiment9,11. Then, Geimet al.11suggested that the local photovoltage created between metal nanoparticles and graphene causes the giant photoeffect. In their plausible mechanism, the generated photovoltage could funnel protons and electrons toward the metal nanoparticles for production of hydrogen, while repel holes away from them.However, this statement is not convincing on the basis of the static electric field theory, as protons and electrons have the opposite charge, and there is no further explanation about it.Otherwise, the microscopic mechanism analysis of this effect is missing in their work11.
Here we demonstrate a microscopic mechanism to explain the origin of the giant photoeffect in proton transport through the catalytically-activated graphene membrane. The local electric fields created between graphene and the metal nanoparticles induce dipole moments of electron orbits nearby. Upon photoexcitation, the local dipoles function as metastable electron states to trap hot electrons with relatively longer life time for effective reaction with injected protons to produce hydrogen.Herein, the catalyst increases the rate of a reaction by increasing the number of successful collisions of reactants instead of lowering the activation energy as usually expected.
2 Results and discussion
Let’s begin our discussion from the origin of the steady proton current. The 100% Faradaic efficiency reported in the experiment11implies that the steady current comes from the chemical reaction of electrons and protons on the other side of graphene. That is, there is no steady current without the chemical reaction. In the case of no chemical reaction, the bias voltage leads to a proton drift current from the proton source through graphene; while the different proton concentration causes an inverse proton diffusion current from the other side of graphene to the proton source. These two currents cancel each other out to reach equilibrium. So, there is no observed net steady current.The net proton diffusion current from the proton source through graphene produce when the chemical reaction occurs because chemical reaction reduces the proton concentration on the other side of graphene. The charge concentration on the other side of graphene should remain unchanged when a steady current is maintained, which gives

wherejpandjeare the current densities of protons and electrons respectively,Rep is the reaction rate of electrons and protons per unit area andqis the absolute charge of a proton or an electron.Because the chemical reaction has an energy barrier, only hot electrons with energy higher than the barrier energy can take part in the chemical reaction. The reaction rate is proportional to concentrations of protons and electrons that can take part in the chemical reaction:Rep=repnp, wherepandnare concentrations of protons and hot electrons respectively, andrepis the intrinsic reaction coefficient. At equilibrium, the proton concentration can be tuned by the bias voltage, while the hot electrons concentration can be influenced by the incident photons.However, for the semi-metal pristine graphene, which has a continuous energy spectrum, most hot electrons excited by photons relax to lower energy states within a timescale of 10-12s16, while the typical timescale of a chemical reaction is 10-6s17,18.In other words, most hot electrons excited by the incident photons will relax to lower energy states before reacting with protons through graphene. This is the reason why no enhancement of the proton current under illumination was observed for graphene without decorating Pt nanoparticles in the experiment11.
In a macroscopic surface potential model, the surface electric field and free charge distribution are simplified to an abrupt model,i.e.the surface electric field is taken as a constant and the free charge is supposed to have a smooth even distribution,resulting a surface space charge region in a semiconductor19.However, many first principle calculations20–24and theoretic models show that the free electrons in both the surface space charge region and the interface specific region (ISR)have modulated oscillatory density distributions rather than even distributions25. The interaction force between graphene and metal nanoparticles is electrostatic force. For graphene-metal equilibrium separation, electrons transfer from one to the other to equilibrate the Fermi levels because the work function of graphene differs from that of metal. This electron transfer results in the formation of interface dipole layer and an accompanying potential step. The local dipoles would be affected by the type and magnitude of the interaction force, because it is induced by the electric field of interfacial space charge layer. The lifetime of a hot electron is determined by the energy level of its ambient which is influenced by interfacial interaction forces. In the case of decorating Pt nanoparticles on graphene, the electrostatic potential at the contact area between Pt nanoparticle and graphene is different from that at the graphene surface without Pt nanoparticle. There are generally two classes of graphenemetal interfaces characterized by the binding energy between graphene and metal. Graphene forms strong interaction with the metal when the binding energy is greater than 2 eV, such as Ni,Pd, Pt, Co, Ru. For the binding energy less than 2 eV, such as Au,Ag, Cu, Ir, graphene forms weak interaction with these metals.When graphene contacts with metal by strong interaction26–31,the interfacial charge redistribution is not only the result of the electron transfer between the metal and the graphene levels. And the graphene-metal strong interaction plays a vital role in the formation of local dipoles as well21. For instance, the giant photoeffect was also observed for graphene decorated with other catalytically active metals Pd and Ni32known to strongly interact with graphene similar to Pt. In contrast, there is no such an effect when Au nanoparticles are used due to its weak interaction with graphene11. Hence, the graphene band structures are strongly perturbed and acquire a mixed graphenemetal character, leading to a modulated non-monotonic oscillatory potential in the ISR (Fig. 1)25,33. Herein, protons are trapped around the negative poles of the local dipoles while electrons are around the positive ones. Upon illumination,electrons are excited to metastable excited states with higher energy levels. Because of energy barriers around them, the free electrons at metastable excited states will have relatively longer lifetime which makes them possible to react with protons before relaxing to ground states. The local surface dipoles in the ISR serve as both proton collection centers and electron storage centers, and finally form chemical reaction centers. However,without illumination the electron concentration at the reaction center is low because of the high electric energy potential there.This leads to a small dark current as observed in the experiment11.

Fig. 1 Schematic illustration of the plane-averaged difference electron density Δn along the z direction showing the local dipoles formed in the specific region of the graphene-Pt nanoparticle interface. Protons are trapped around the negative poles of the local dipoles while negative free charges are near the positive ones. Electrons excited to metastable excited states with higher levels by photons can react with protons moreeffectively due to a relatively longer life time.
Under illumination the electrons at the ground state are transited to the metastable excited states with higher energy by the electric field of photons. Transitions between different metastable excited states may also happen. To simplify the discussion we employ an effective two level model to describe this phenomenon, with the framework showed in Fig. 2. The effective ground state is labeled asand the effective excited state is labeled asThe energy difference between the excited state and the ground state isEe. The mean effective energy of photons isħω, whereωis the mean effective angular frequency of photons. Generally there is an energy detuning between the energy difference of the two electron states and the photon energy:δ=Ee–ħω. The photon interacts with the electronviaits electric field:whereis the dipole moment of the electron andis the electric field of the photon. The whole Hamiltonian can be simplified to the Jaynes-Cummings (JC)model by eliminating the high frequency part34:

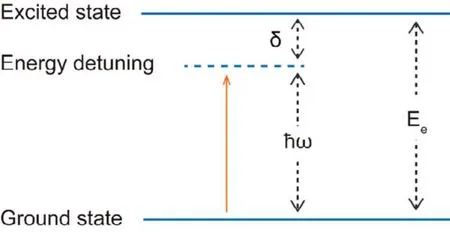
Fig. 2 Framework of the effective two level model.
whereΩis the photon-electron coupling constant,andare creation and annihilation operators of photons respectively,The Hamiltonian can be solved in a subspacewheremis the photon number density in grapheneThe eigenstates and eigenvalues of the JC model are:

Because of the heating effect of illumination, the effective temperature of electrons under different illumination strength varies from each other and it is hard to predict the exact state of electrons without the knowledge of the exact temperature. Here we first resort to a very rough estimate of the excited electron state occupation by assuming a zero environment temperature and only the lowest eigenstate of the whole interacting Hamiltonian is occupied:

The concentration of high energy electrons is estimated aswheren0 is the effective total electron concentration,Iis the power density of the incident photon current andI0=Iδ2/(m+ 1)Ω2=ħωδ/Ω2is supposed to be a constant parameter. The equation shows that more electrons are pumped to the excited state under stronger illumination. The proton current density thus reads:
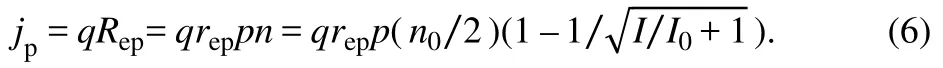
For weak illuminationI< while for strong illuminationI>>I0the proton current density saturates at: With finite environment temperature the concentration of high energy electrons is corrected as, wherekBis the Boltzmann’s constant,Tis the Kelvin temperature. The proton current density becomes: Leta=qreppn0/2,b=I0 andc=ħ δ/2kBTbe three fitting parameters, the fitting curves are shown in Fig. 3, indicating a reasonable agreement with the experiment data of the proton current density11although very rough approximations are made in the model. For weak illumination the heat effect is negligible and the temperature is invariant. While for strong illumination the proton current is underestimated by the model because of the heavy thermal effect. It should be noticed that the interface electric field distribution and the induced local dipole as described in the propose model could be impacted when metal particles have different sizes due to the quantum confinement effect. Fig. 3 Proton current density as a function of illumination power for different biases. Researchers have been attempted to take good advantage of hot carriers in photo(electro)catalysis, but the role is still not so significant35–39. On the basis of the above analysis, the giant photoeffect in proton transport through the catalyticallyactivated graphene membrane is attributed to long lived hot carries and fast proton transport rate. This reasoning is supported by many works. Since graphene is a strictly two-dimensional zero-bandgap semiconductor with a liner energy dispersion, it is theoretically predicted that the impact of carrier multiplication on photocurrent response might be enhanced by very inefficient electron cooling, resulting in an abundance of hot carriers40–42.Indeed, long lived hot carries have been reported in many experiments for graphene with built-in p–n junctions43–45,explained as photovoltaic16,46–48or photothermoelectric effect49,50.On the other hand, the rate for proton permeation through graphene is of the order of 1013–1014s-13, about 100 times faster than the lifetime of hot electrons, which has enough number to fully react with hot electrons for producing hydrogen. Taken together, the local dipoles induced by graphene and the metal nanoparticle function as metastable electron states with higher energy to trap hot electrons with relatively longer life time upon photoexcitation for effective reaction with large amount of injected protons to produce hydrogen. Since there is no change in the activation energy of the reaction, the metal catalyst increases the rate of a reaction by increasing the number of successful collisions of reactants for this significant photoeffect.This deduction is corroborated by the claim that the ratedetermining factor in the photocatalytic system is the concentration of surface-reaching photocharges before it reaches the threshold, instead of the widely accepted speculation of high reaction barriers to be overcome24,51. In most practical photo(electro)catalytic systems, the rapid photogenerated charge recombination52,53and possible inhibit of mass transfer due to the complex electric double layer or ions in solution54–56cause inferior performances. In summary, we developed a microscopic mechanism about the new role of the catalyst based on the experiment of the illumination enhancement of proton transport through catalytically-activated graphene. Hot electrons in pristine graphene cannot participate a chemical reaction because of their short lifetime. When catalytically-active metal nanoparticles are decorated on graphene, they induce local dipoles in the interface specific region. Electrons can be trapped around the positive poles of the local dipoles, and protons are trapped around the negative ones after permeating through graphene. Upon illumination, electrons at the ground state can be pumped to metastable states with longer lifetime, so the chemical reaction between hot electrons and protons are accelerated. Our simplified two level model estimates the occupation of the excited electron state, and the corresponding fitting curves of the proton current density as a function of the incident light power density agree reasonably well with the experiment data. In the new mechanism the effect of metal nanoparticles is to stimulate local dipoles which provide long lived metastable excited states of electrons, instead of lowering the activation energy. A simplified effective two level model is also provided to explain the photocurrent density behavior under different illumination power density. This mechanism may shed light on the microscopic catalytic process and provide a guidance in designing photoelectrochemical energy conversion materials or devices. Furthermore, this mechanism can be generalized to other interfaces with the electrostatic interaction. Different from metal and semimetal systems, the lifetime of free electrons in semiconductor systems will be not extended by the formation of new metastable excited states because the conduction band edge of a semiconductor is actually a stable excited state. However,the function of local dipoles can be reflected in some microscopic and transient phenomenon such as photoluminescence spectra and the lifetime of photoluminescence.



3 Conclusions
