Personalized immunotherapy in cancer precision medicine
2021-11-20KazumaKiyotaniYujiroToyoshimaYusukeNakamura
Kazuma Kiyotani, Yujiro Toyoshima, Yusuke Nakamura
Project for Immunogenomics, Cancer Precision Medicine Center, Japanese Foundation for Cancer Research, Tokyo 135-8550,Japan
ABSTRACT With the significant advances in cancer genomics using next-generation sequencing technologies, genomic and molecular profilingbased precision medicine is used as a part of routine clinical test for guiding and selecting the most appropriate treatments for individual cancer patients. Although many molecular-targeted therapies for a number of actionable genomic alterations have been developed, the clinical application of such information is still limited to a small proportion of cancer patients. In this review, we summarize the current status of personalized drug selection based on genomic and molecular profiling and highlight the challenges how we can further utilize the individual genomic information. Cancer immunotherapies, including immune checkpoint inhibitors,would be one of the potential approaches to apply the results of genomic sequencing most effectively. Highly cancer-specific antigens derived from somatic mutations, the so-called neoantigens, occurring in individual cancers have been in focus recently. Cancer immunotherapies, which target neoantigens, could lead to a precise treatment for cancer patients, despite the challenge in accurately predicting neoantigens that can induce cytotoxic T cells in individual patients. Precise prediction of neoantigens should accelerate the development of personalized immunotherapy including cancer vaccines and T-cell receptor-engineered T-cell therapy for a broader range of cancer patients.
KEYWORDS Personalized medicine; cancer precision medicine; neoantigen; personalized immunotherapy; immune checkpoint blockade; cancer vaccine; adoptive T cell therapy
Introduction
In the past decade, cancer treatment has been significantly improved toward precision medicine on the basis of individual genomic information. The advances in next-generation sequencing (NGS) technologies offer us opportunities to obtain a comprehensive cancer genome landscape, including genetic alterations, gene expression, and epigenetic profiles. A personalized approach based on the genome information of individuals’ cancer has potentials to identify clinically actionable target molecules and is useful in selecting appropriate treatments for individual cancer patients. These genomic-based cancer diagnostics and treatment are becoming a standard procedure. Indeed, several NGS-based cancer diagnostic tests,in addition to conventional sequencing- or PCR-based tests,have been approved by the US Food and Drug Administration(FDA). These cancer diagnostic tests can serve as companion diagnostics for approved molecular-targeted drugs and are utilized for patient enrollment in clinical trials of targeted cancer therapies. However, a small subset of patients can receive benefit from the genetic analysis because the number of molecular-targeted drugs is still limited. Immunotherapy is one of the novel treatment modalities that include the checkpoint blockade therapy, personalized cancer vaccines, and adoptive T-cell therapies. In this review, we will discuss the current status and future direction of implementing cancer precision medicine in the clinical setting, specially focusing on the personalized immunotherapies.
Somatic mutation-based selection of molecular-targeted drugs
Genetic or molecular profiling of individual tumors would provide critical information to predict efficacy and/or risk of toxicity of drugs. With the recent advances in sequencing technologies, many genetic biomarkers including somatic and germline mutations, gene amplifications or fusions have been identified. A large-scale meta-analysis of 346 phase I clinical trials clearly demonstrated that biomarker-based drug selection was significantly correlated with a higher response rate of 30.6% compared to 1.9% in the non-personalized treatment group1. The mutational landscape of metastatic cancers of more than 10,000 patients with clinical sequencing showed that up to 80% of tumors sequenced by the NGS-based targeted gene panel tests had at least one genetic alteration2.Approximately 40% of the patients who were subjected to the NGS tests had one or more potentially actionable alteration;however, only 10%-15% ended up being treated with genotype-guided appropriate drugs because of declining patients’performance status, limited accessibility to clinical trials or limited availability of molecular-targeted drugs (Figure 1 and Table 1)2-6. Based on these studies, 2 cancer profiling tests,MSK-IMPACT and FoundationOne CDx, examining the panels of genetic alterations in 468 and 324 cancer-associated genes using NGS technology were approved by the US FDA in 2017. In the patients enrolled to these genotype-matched clinical trials, the objective response rates of these treatments were as modest as 20% or less (only 2%-3% of patients who were sequenced in the clinical trials), which was probably due to the entry of advanced-stage cancer patients after failure of at least one line of standard therapy, but the rates were significantly higher than those in the unmatched therapy group.
Compared with targeted gene-panel sequencing, whole-genome sequencing (WGS) and whole-exome sequencing(WES) provide us more comprehensive genomic profiles of individual tumors. A recent study that analyzed 2,520 samples in 22 types of metastatic tumors by WGS demonstrated that 62% of these tumors contained at least one actionable mutation7. In another study, of 62 patients with no actionable finding in a targeted gene-panel testing, WES and RNA sequencing (RNAseq) identified one or more known cancer driver events in 25 (40%) patients8. WES/WGS and RNAseq improved detection rates of actionable biomarkers or oncogenic mutations, but their impacts on cancer patients to provide clinical benefits are still very limited. Therefore, further development of novel molecular-targeted drugs and other treatment options is urgently needed.
Immune checkpoint inhibitors
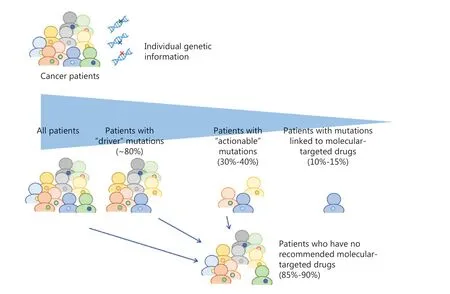
Figure 1 Summary of frequency of actionability and patients’ response rates in genomic profiling-based clinical trials. Approximately 40%of the patients had potentially actionable mutations/alterations, but only 10%-15% were treated with genotype-guided appropriate drugs.
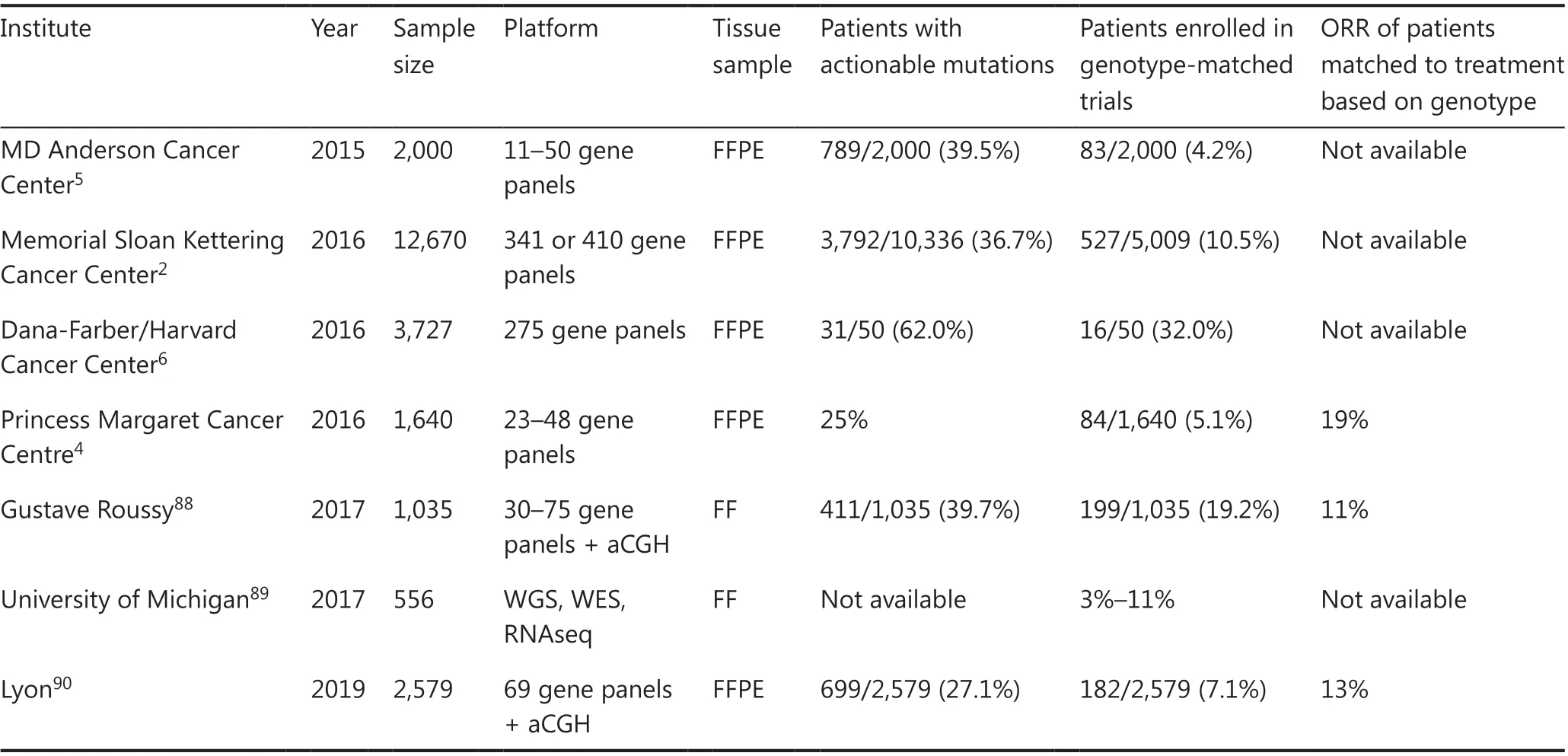
Table 1 Selected clinical trials of genotype-based therapy
T cells infiltrate tumor sites to eliminate cancer cells, but these T cells are often suppressed by immunosuppressive molecules and cells. Inhibitory checkpoint molecules expressed on T cells, including program death 1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), are typical key negative regulators of T cell-mediated immune response and therefore are considered as targets for cancer immunotherapy. Immune checkpoint inhibitors (ICIs) such as antibodies against PD-1, its ligand PD-L1, and CTLA-4 block these immune inhibitory molecules and restore the anti-tumor activity of cytotoxic T cells, resulting in eradication of cancer cells. ICIs have drastically improved cancer treatments, and to date, 7 ICIs, namely, nivolumab, pembrolizumab, and cemiplimab for PD-1; avelumab, atezolizumab, and durvalumab for PD-L1; and ipilimumab for CTLA-4, were approved by the US FDA. Despite ICI therapy showing significant clinical benefits in patients with various types of cancer, only 20%-30%of the patients have shown clinical responses, and a majority of patients experienced no or limited clinical benefit9. These lines of evidence indicate the importance to identify a biomarker(s) to predict patients’ clinical benefit and the risk of immune-related adverse events, contributing to an increase in patient safety and decrease in unnecessary medical costs9.Many reports have indicated that a higher level of CD8+T-cell infiltration was associated with better clinical responses to ICI therapy10. PD-L1 expression in both cancer cells and immune cells was associated with clinical responses; therefore,detection of PD-L1 expression has been used as a companion diagnostic testing for PD-1/PD-L1 checkpoint inhibitors10. Contrarily, PD-L1 expression levels in the tissues do not consistently correlate to therapeutic responses to anti-PD-1/PD-L1 therapies11. This inconsistency may be explained partly by the effects ofN-linked glycosylation of PD-L1, which might interfere with the binding of clinically used anti-PD-L1 antibodies. Prior deglycosylation of cancer tissues altered the PD-L1 status from negative to positive in approximately 16% of tumors in responders to anti-PD-L1 therapy, and the PD-L1 expression scores were significantly correlated to overall survival (OS) after but not before the deglycosylation (P=0.005 andP= 0.29, respectively) in a study of 95 patients who received anti-PD-L1 therapy12. Therefore, deglycosylation of PD-L1 might be an effective method to improve the accuracy of PD-L1 detection as a biomarker for immune checkpoint therapy, although further validation in larger-scale clinical trials is necessary to apply into the clinical setting.
PD-L1 genomic amplification was observed in 843 (0.7%) of 118,187 patients with various types of cancer13. Among them, 6 of 9 patients with genomic amplification of PD-L1 revealed an objective response to ICIs; therefore, the PD-L1 amplification may be useful as a predictive biomarker13. Genomic, transcriptomic, and other technologies have been employed to identify a biomarker(s) that predicts responses to the ICI therapies. One of possibly useful biomarkers to predict checkpoint inhibitor responsiveness indicated by genomic approaches is the number of somatic mutations altering amino acid sequences, called as tumor mutation burden (TMB), that lead to the generation of tumor-specific antigens known as neoantigens14-16. In an analysis of 1,638 immunotherapy-treated patients, the higher(≥20 mutations/Mb) TMB was an independent predictor for better progression-free survival (PFS) and OS14. Another analysis of 1,662 advanced cancer patients treated with ICIs also showed that patients with higher TMB (highest 20% in each cancer type) had significantly better OS16. However, it is still difficult to define one universal cutoff threshold of high TMB;thus, harmonization initiatives, named as tumor mutational burden standardization initiatives, are now standardizing methods to detect TMB for diagnostic uses17.
High microsatellite instability (MSI-H) resulting from defects in DNA mismatch repair genes, in turn leading to higher TMB with > 1,000 non-synonymous mutations, which is 10 times or higher than the number of mutations in microsatellite stable (MSS) tumors, is an established biomarker for prediction of ICI responses18,19. Based on the findings, some ICIs have been approved by the US FDA for the treatment of patients with MSI-H tumors, irrespective of an origin of organs20-23. In addition, somatic mutations in polymerase δ(POLD1) and polymerase ε (POLE) genes, which lead to an ultramutator phenotype (very high TMB), were associated with high infiltration of immune active cells into tumor sites and better responses to ICIs15,24,25. Meanwhile, loss-of-function mutations/alterations in genes related to an antigen presentation machinery and an interferon (IFN) signaling pathway (B2M,HLA,JAK1,andJAK2) were associated with poor responses to ICIs26.PTENloss was shown to promote resistance to PD-1 inhibitor therapy in melanoma patients by increasing the expression of immunosuppressive cytokines, resulting in decreased T-cell infiltration in tumors27. This result suggests that combination of inhibitors of the PI3K/AKT/mTOR pathway may improve the efficacy of immunotherapy27. Mutations inEGFRwere also reported to associate with lower response rates to ICIs in patients with lung cancer probably due to low TMB leading to immunosuppressive microenvironment28,29.In addition,STK11mutations were reportedly associated with resistance to immunotherapy30. However, the available genomic biomarkers still do not adequately predict response to immunotherapy. Gene expression signatures defined by transcriptome analyses may be useful to predict the tumor responses to immune checkpoint therapies. It was reported that the T-cell-inflamed gene expression profile consisting of 18 IFN-γ-responsive genes related to antigen presentation,chemokine expression, cytotoxic activity, and adaptive immune resistance exhibited predictive utility in identifying responders to anti-PD-1 and anti-PD-L1 therapies, regardless of tumor types in concordance to the data based on IHC31. In addition to PD-L1 expression, we found that the higher expression of granzyme A (GZMA) and HLA-A was observed in pretreated tissues of melanoma patients who responded to anti-PD-1 therapy, which can be potential biomarkers to expect clinical responses32. Furthermore, we identified a clonal enrichment of tumor-infiltrating T cells with certain T-cell receptor (TCR)repertoire in responders32. Recently, it has been suggested that TCR repertoire dynamics in patients’ peripheral blood at an early phase of the treatment may be useful to predict or monitor the response and resistance to ICIs33-35. We found that sustained expansion of certain dominant T-cell clones was detected in peripheral blood in patients with clinical response to ICI therapy in lung and kidney cancer33-35.
Neoantigens
Neoantigens are highly cancer-specific antigens generated by somatic mutations in cancer cells and therefore are not present in normal cells36. Due to their high cancer specificity, neoantigens are considered as promising targets of cancer immunotherapy. Although neoantigens were first studied during the late 1980s using mouse models37,38, they have been paid a significant attention in recent years because of recent remarkable advances in NGS, which provides us comprehensive genomic landscape of tumors. The associations of high TMB resulted in high neoantigen load with the high intratumoral T-cell infiltration have been well studied in cancer patients who received not only ICIs but also other treatments such as chemotherapy and/or radiation therapy. TMB and predicted neoantigen load were significantly correlated with cytolytic activity estimated by the expression of key immune effector molecules,GZMAand perforin 1 (PRF1), that are highly expressed in activated cytotoxic T lymphocytes (CTLs) in large-scale WES and transcriptome data sets across 18 cancer types and were also significantly associated with better patient prognosis in 515 patients across 6 cancer types in The Cancer Genome Atlas39,40. These associations of higher neoantigen load with high T-cell infiltration into tumors and better clinical outcomes were supported by many studies in various cancer types, including both MSI and MSS tumors41,42. We found that patients with DNA repair gene mutations, resulting in higher TMB, showed significantly better recurrence-free survival than patients without DNA repair gene mutations (hazard ratio, 0.46;P= 0.044) and that an oligoclonal expansion of tumor-infiltrating T cells, represented by lower TCR diversity, was observed in tumors with higher neoantigen load in WES data of 78 patients with muscle-invasive bladder cancer43,44. We also demonstrated that a higher neoantigen load was significantly correlated with lower TCR diversity in malignant mesothelioma and breast cancer45,46.Furthermore, higher neoantigen load and lower T-cell diversity were significantly associated with clinical responses to neoadjuvant chemoradiotherapy in rectal cancer47,48.
Since every cancer has its own unique mutations, neoantigens that possibly induce CTLs targeting cancer cells are also different in individual cancer patients. However, because it is hard to know the presence or absence of neoantigen-specific T cells in each patient, it is still challenging to accurately predict neoantigens that actually induce CTLs in patients by several pipelines currently available for prediction of neoantigens that are basically predicting the binding affinity of neoantigen peptides to HLA molecules. We have developed a neoantigen prediction pipeline using WES and/or RNAseq data, which consists of the following 4 steps (Figure 2)49. The first step isHLAgenotyping based on WES data. Since cytotoxic CD8+T cells recognize antigen peptides presented on HLA class I moleculesviatheir TCR, at least 2-field HLA class I genotyping, which classifies different protein sequences, is required for neoantigen prediction. In our pipeline, we applied OptiType and Polysolver, which showed high accuracies of 97.2% and 94.0%, respectively, for HLA class I alleles in 961 WES data from the 1000 Genomes Project50-52. The second and third steps are variant call and detection of mutant RNA expression.Somatic single nucleotide variants (SNVs) as well as insertions/deletions (indels) are identified by comparing exome data from tumors and matched normal samples, and annotation was added to each mutation by Annovar53. We target non-synonymous SNVs and indels, which alter amino acid sequences. Among these SNVs and indels, we identify mutated peptides expressed in cancer cells by separately counting the reads of wild-type and mutant RNA using tumor RNAseq data. This step is particularly important to accurately select neoantigen candidates because mutated RNA expression was not always correlated with total RNA (wild-type + mutant RNA) expression obtained as FPKM or RPKM54. Thus far, we have found that approximately 45.0% (ranging from 11.1% to 72.7%) of mutated genes were expressed in each cancer tissue45,46. Although a very low level of antigens, as low as a single peptide on HLA/cell, is sufficient for activation of CTLs55,the appropriate cutoff for RNA expression for selecting neoantigens that can effectively activate CTLs remains debatable.The last step is prediction of the binding affinity between peptides and HLA molecules. NetMHC and NetMHCpan are the most commonly used prediction programs, which are on the basis of artificial neural networks using large datasets of experimentally validated HLA-binding peptides collected in the Immune Epitope Database (IEDB)56-59. Some other bioinformatic programs, including NetMHCstab, NetChop, and NetCTL, predict peptide-HLA complex stability, antigen processing, and peptide transport processes, respectively60-62.Recently, HLA-peptide affinity prediction tools using several artificial-intelligence approaches have been also reported63-65.Mass spectrometry-based immunopeptidome analysis to identify peptides that are presenting on HLA molecules is one of the approaches to narrow down the candidate peptides,although improvement of sensitivity is critically essential to optimize the potential of immunopeptidome analysis for clinical application66.
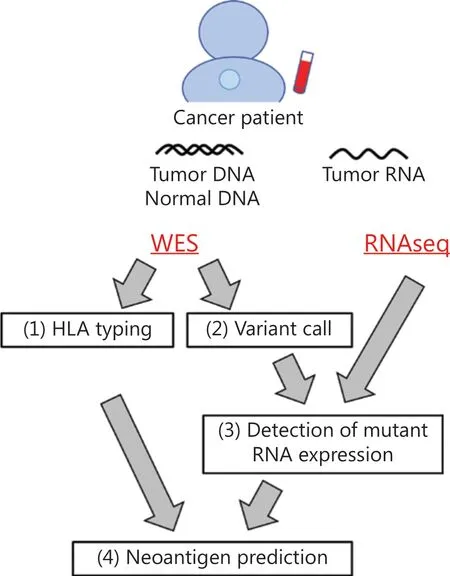
Figure 2 Workflow of neoantigen prediction pipeline from genome sequencing data. The first step is HLA class I genotyping(at 2-field level) based on WES data. The second and third steps are somatic variant call and detection of mutant RNA expression. Nonsynonymous SNVs and indels, which alter amino acid sequences,were selected and further analyzed. The last step is binding affinity prediction of mutated peptides to individuals’ HLA molecules. WES,whole-exome sequencing; RNAseq, RNA sequencing; SNVs, single nucleotide variations; Indels, insertions/deletions.
As described above, due to a significant improvement of bioinformatic tools and accumulation of large experimental data,we were able to predict the interaction between peptides and HLA molecules; however, it is still very difficult to predict the interaction between HLA-peptide complex and TCR; furthermore, at present, we have no methods to know the presence of T cells with antigen-specific TCR in patients. Therefore,experimental validation of predicted neoantigen peptides using T cells from patient’s peripheral blood or tumor-infiltrating T cells is needed to confirm their immunogenicity. We developed an experimental pipeline to induce neoantigen-reactive T cells from donors’ or patients’ peripheral blood and to identify TCRs specifically recognizing neoantigens within approximately 2 weeks from the peptide stimulation of T cells to the identification of specific TCR67-69. Using this approach,we successfully induced several neoantigen-reactive T cells and identified TCR sequences of these neoantigen-reactive T cells. There are several reports investigating the induction of neoantigen-reactive T cells from tumor-infiltrating T cells,peripheral blood of patients or healthy donors, using short neoantigen peptides, long peptides, or tandemly connected minigenes70. Overall, 5%-20% of neoantigens predicted byin silicoprograms induced neoantigen-reactive T cells inin vitroinduction experiments. Further accumulation of big data of immunogenic peptides and improvement of the prediction accuracy will be required. Importantly, during the screening, we found that neoantigen-reactive TCRs are not always specifically reactive to mutated neoantigen peptides, but are also reactive to corresponding wild-type peptides in some cases68. We clarified that the position of mutated amino acids within the peptide sequence is an important determinant to predict the cross-reactivity against wild-type peptides67-69.Therefore, structure-based approach may be useful to predict the cross-reactivity. These data are critically important to avoid the risk of life-threatening adverse events. In addition to the property and immunogenicity of neoantigen peptides, the characteristics of tumor, such as intratumoral heterogeneity and immune-related gene expression, need to be considered to select more effective neoantigens. Since loss of antigen presentation is one of the most common immune evasion mechanisms, targeting neoantigens derived from clonal driver mutations, which are important for tumor growth, is crucial.Information pertaining to mutations and loss of heterozygosity of theHLAgene is also needed for effective neoantigen candidate selection51,71Regarding the loss ofHLAexpression,if it is caused by epigenetic changes inHLAgenes, the combination with the epigenetic modifiers might help to increase the expression levels of HLA molecules.
Personalized immunotherapy targeting cancer-specific neoantigens
In recent years, several cancer immunotherapies have been extensively studied to enhance anti-tumor immune responses mediated by CTLs. Adoptive transfer therapy of tumor-infiltrating T-lymphocytes (TILs), in which autologous T cells infiltrated into tumor are harvested, expandedin vitro, and administered to the patient, showed promising results especially in melanoma patients72,73. In a clinical trial of TIL-based adoptive transfer therapy in metastatic melanoma patients who had failed their standard regimens, complete responses(CRs) were reported in 20 (22%) of 93 patients, and the objective response rate was 56% irrespective of prior therapy(ies)72.Toxicities from the therapy were predominantly associated with lymphodepletion and high-dose interleukin 2 (IL-2),including grade 3 toxicities of neutropenia, anemia, thrombocytopenia, febrile neutropenia as a consequence of lymphodepletion, capillary leak syndrome, and hyperbilirubinemia.Several clinical trials are ongoing to assess the efficacy of TIL therapy in various solid tumors.
Cancer vaccines, including peptide vaccine and dendritic cell vaccine, targeting neoantigens or shared antigens, induce and activate CTLs reactive to these antigens in cancer patients.The results of many clinical trials indicate the clinical utility of peptide vaccines targeting shared antigens derived from oncogenes specifically expressed in cancer tissues, oncoantigens74-76. In our recent phase II trial for adjuvant cancerspecific oncoantigen peptide vaccine for esophageal cancer patients who had neoadjuvant chemo(radiation)therapy and curative resection, but were found to have lymph node metastasis, patients treated with peptide vaccine showed a significantly higher 5-year esophageal cancer-specific survival rate than the non-vaccinated group (60.0%vs32.4%,P=0.045); the difference was more significant in patients with tumors without CD8+or PD-L1 expression (68.0%vs17.7%,P= 0.010)76. Several phase I clinical trials have been conducted to evaluate the efficacy of neoantigen-targeted personalized cancer vaccines (Table 2). In a study of 13 melanoma patients who received RNA-based neoantigen vaccine,all patients developed T-cell responses against neoantigens,and the cumulative rate of metastatic events was significantly reduced by the vaccination, resulting in a sustained PFS77. In another study of 6 melanoma patients who received vaccine treatment that targets up to 20 predicted personal neoantigens, 4 patients had no recurrence for nearly 2 years after the beginning of vaccine treatment and 2 patients showed CR with expansion of neoantigen-specific T cells in combination with anti-PD-1 therapy78. Two neoantigen-based cancer vaccine trials were also conducted for glioblastoma (Table 2)79,80.Hilf et al.80investigated neoantigen- and/or shared antigen-based peptide vaccines in 15 glioblastoma patients and found that one patient showed partial response (PR) and the other revealed stable disease (SD) and that the median PFS and OS were 14.2 and 29.0 months, respectively. Keskin et al.79showed that the neoantigen vaccine was a feasible therapeutic strategy for glioblastoma with low TMB; however, all patients had progressive disease with a median PFS of 4.6 months and OS of 16.8 months. Recently, Ott et al.81conducted a clinical trial for personalized neoantigen vaccines with anti-PD-1 antibodies in 60 patients with melanoma, non-small cell lung cancer (NSCLC), or bladder cancer and found that neoantigen-specific CD4+and CD8+T-cell responses were observed in all of the patients and that objective response rates were 59%, 39%, and 27% for melanoma, NSCLC, and bladder cancer, respectively. In these clinical trials, the most frequently observed adverse events were grade 1-2 injection site reactions and influenza-like illness, including fatigue, chills, and fever, and grade 3 treatment-related toxicities were observed only in less than 10% of patients. These results have indicated that neoantigen vaccines designed based on individual genomic information can be one of the promising approaches to induce cancer-reactive CTLs in cancer patients.
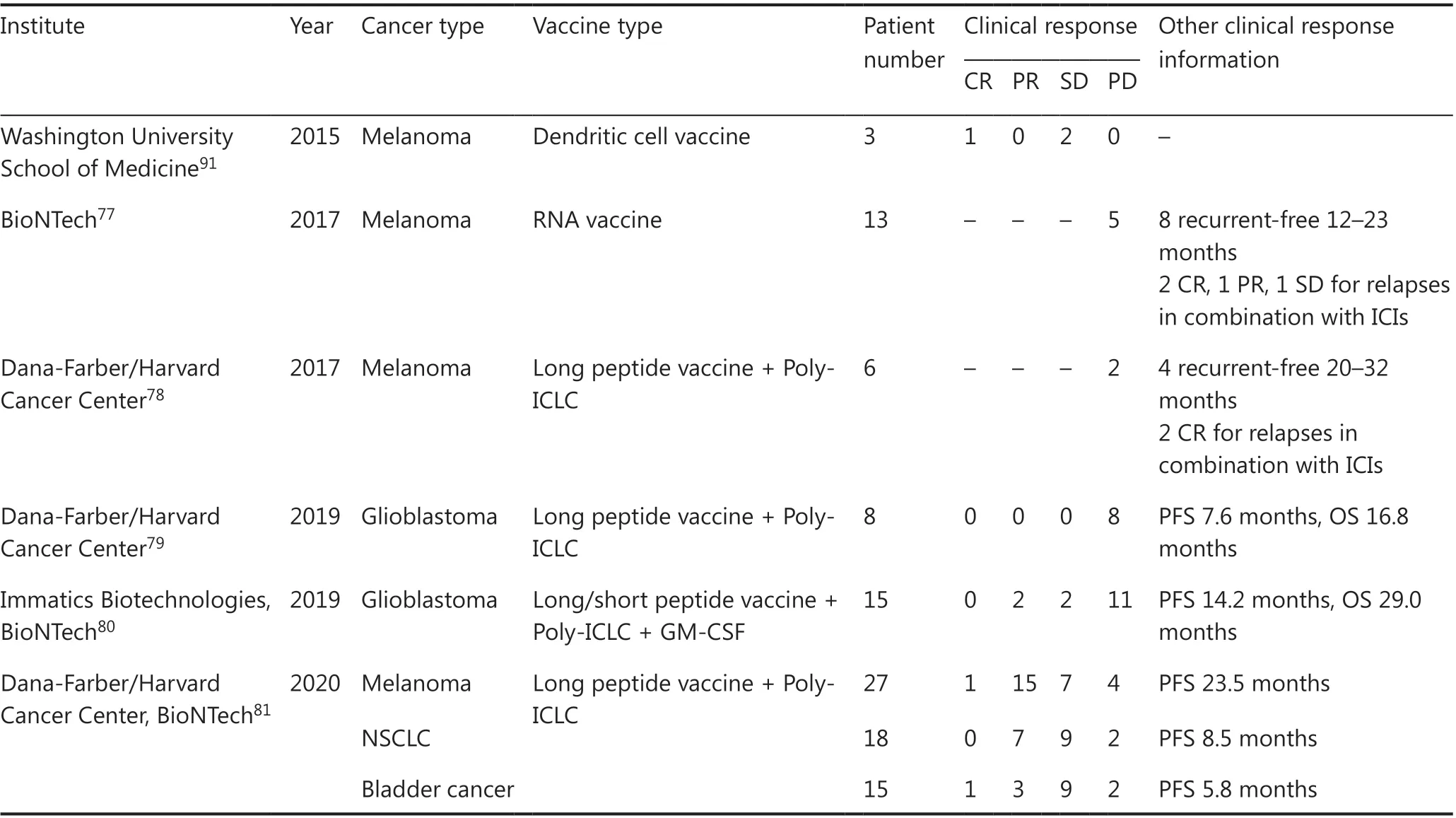
Table 2 Published clinical trials of personalized neoantigen vaccines
In addition of cancer vaccine therapy, adoptive T-cell transfer therapy using TCR-engineered T (TCR-T) cells have been investigated82. Compared with adoptive therapy using patients’ TILs, this approach has the advantage of continuously supplying cancer-reactive T cells and effectively inducing anti-cancer CTL responses even in tumors in advanced stage where the host immune system is extensively suppressed.TCR-T-cell-based adoptive cell therapy was first reported for shared antigen MART-1 in 200683. In the study, 2 of 15 metastatic melanoma patients who received adoptive transfer of genetically engineered T cells showed objective regression of the metastatic melanoma lesions. Adoptive T-cell therapy using TCR-T cells targeting NY-ESO-1 has been most widely investigated, and it was reported that objective response rates were 55%, 61%, and 80% in patients with melanoma, synovial cell sarcoma, or multiple myeloma, respectively, without severe side effects, although all patients experienced the transient toxicities associated with high-dose of IL-284,85. Based on these results, multiple clinical trials are now ongoing to confirm the efficacy of NY-ESO-1-specific TCR-T-cell therapy in various types of solid tumors. Although no report has been published on adoptive transfer therapy of personalized TCR-T cells targeting individual neoantigens in humans, the effectiveness of neoantigen-specific TCR-T cells has been shown to eradicate large-size solid tumors in mouse models86,87. We found that a single adoptive transfer of TCR-T cells targeting p68 mutation, S551F, eradicated an established solid tumor with a diameter of 1 cm carrying the p68 mutation86.With all the findings so far, personalized cancer vaccines and adoptive T-cell therapy-targeting neoantigens might provide us a possibility of ultimate personalized treatments for cancer patients.
Conclusions
In this review, we mainly focused on the current status of personalized immunotherapies including neoantigen-based cancer vaccine and TCR-T-cell therapies, which are highly specific to cancer. Personalized immunotherapy could provide novel and alternative treatment options for patients who do not have actionable mutations for molecular-targeted drugs or become resistant to conventional chemotherapy. Although further improvement in the accuracy to select target neoantigens to individual patients is still critically essential, personalized neoantigen-targeting immunotherapies have the possibility of becoming an ultimate precision cancer treatment.
Grant support
This work was partly supported by Japan Agency for Medical Research and Development (Grant Nos. 17ck0106364h0003 and 20ck0106543h0001) and by the Japan Society for the Promotion of Science (Grant No. 19H03522).
Conflict of interest statement
Kazuma Kiyotani is a scientific advisor of Cancer Precision Medicine, Inc. Yusuke Nakamura is a stockholder and a scientific advisor of OncoTherapy Science, Inc.
杂志排行

Cancer Biology & Medicine的其它文章
- Insights into tertiary lymphoid structures in the solid tumor microenvironment: anti-tumor mechanism, functional regulation, and immunotherapeutic strategies
- Culturing adequate CAR-T cells from less peripheral blood to treat B-cell malignancies
- EZH2 identifies the precursors of human natural killer cells with trained immunity
- The role of DLL1 in long-term vascular normalization and cancer immunotherapy
- Multiplex imaging reveals the architecture of the tumor immune microenvironment
- The commensal consortium of the gut microbiome is associated with favorable responses to anti-programmed death protein 1 (PD-1) therapy in thoracic neoplasms