生物催化轴手性化合物的不对称合成进展
2021-11-12尚亚莉
尚亚莉, 袁 波, 费 强
生物催化轴手性化合物的不对称合成进展
尚亚莉, 袁 波, 费 强
(西安交通大学 化学工程与技术学院, 陕西 西安 710049)
轴手性化合物是许多天然产物、药物中间体、手性配体的核心骨架,在手性化合物中占有重要地位。与金属催化剂催化的不对称化学偶联反应等化学方法相比,生物催化方法具有选择性高、反应条件温和、环保等优势。随着酶的改造等关键技术的快速发展,酶催化轴手性化合物的合成成为新的研究热点与难点。从动力学拆分(kinetic resolution,KR)、动态动力学拆分(dynamic kinetic resolution,DKR)以及去对称化(desymmetrization)等不对称合成方法入手,综述了生物催化轴手性化合物的合成领域的主要研究成就,并阐述了此方向的发展前景、应用及存在的问题。
生物催化;轴手性化合物;不对称合成;动力学拆分;动态动力学拆分;去对称化
1 前 言
轴手性是指由于空间位阻或电子约束导致单键旋转受阻从而表现出实体与镜像不重合的现象。当轴的旋转能垒足够大(> 96.27 kJ×mol-1),半衰期足够长(>1000 s)时,轴手性化合物在室温下能稳定存在,并可拆分为2种异构体[1]。高手性纯度的轴手性化合物可通过数种不对称合成方法制备,例如动力学拆分(kinetic resolution,KR)、动态动力学拆分(dynamic kinetic resolution,DKR)以及去对称化(desymmetrization)等。
轴手性化合物在天然产物合成[2-3]、手性配体制备、药物中间体[4]以及功能性材料[5]合成等重要领域皆有广泛的应用。2001年,日本名古屋大学的野依良治(R. Noyori)因在不对称氢化领域的突出贡献获得诺贝尔化学奖,轴手性化合物2,2'-双(二苯基膦)-1,1'-联萘(2,2'-bis(diphenylphosphino)-1,1'-binaphthyl,BINAP[6])则作为金属催化剂手性配体[7-8]在不对称氢化领域发挥着重要作用。含轴手性结构的药物中,万古霉素(vancomycin)被认为是抵抗细菌感染的最后一道“防线”,临床上广泛应用于抗药性细菌引起的重度感染的治疗。其化学全合成由Evans等[9]与Nicolaou等[10]分别完成。万古霉素的核心骨架目前尚无生物合成方法[11]。Knipholone[12-14]的关键结构为轴手性联苯键,其化学全合成中关键的轴手性选择性耦合步骤由“内酯法”达成。生物碱Murrastifoline-F含有手性的C-N轴,其化学合成由加入手性诱导分子并进行色谱分离完成[15]。轴手性化合物的化学合成法目前仍为主要合成方法,其应用范围广且可操作性强。但是,化学法往往需要色谱分离,其量级受到严重限制;或者需要手性诱导分子,其辅助分子需要进一步去除,增加了反应步骤,降低反应效率;并且部分金属催化剂价格高昂且可能对环境造成一定污染。2010年,Science报道了来自耶鲁大学的Gustafson利用手性诱导分子合成具有轴手性的联芳基产物的研究,是化学法合成轴手性底物的一个经典案例[16]。化学法近年来在本领域的飞速发展也带动了生物催化方法的研究,生物法合成轴手性分子具有条件温和、安全环保、立体选择性和区域选择性优良等优点,应用领域仍有待拓展。本研究概述了近年来制备高手性纯度的轴手性化合物的生物催化方法。从酰化反应到氧化还原反应,从联烯类底物到联芳基类底物,从商业酶类到新型酶变体,力图从各个层面较为完整地概述本领域研究进展,为生物催化轴手性化合物合成的研究提供有效参考。
2 生物催化轴手性化合物的动力学拆分(KR)反应
KR是最经典的不对称合成方法之一。包括脂肪酶(lipase)等在内的水解酶(hydrolase)催化的酰化反应是KR反应中最常见的类型。由于脂肪酶稳定性好、获取容易且成本低,使其应用极为广泛。而氧化还原酶亦可用于轴手性化合物的KR反应,但通常需要价格昂贵的辅酶或辅因子。近年来随着辅酶回收技术的发展,氧化还原酶的研究得到快速发展。本章节主要介绍生物催化轴手性化合物的KR反应,其中对脂肪酶催化的酰化反应和氧化还原酶催化的氧化还原反应展开重点分析。
2.1 酰化反应
酰化反应是指有机物中的C、N、O等原子上导入酰基的反应。酰卤、酸酐、羧酸、羧酸酯等都可以作为酰化剂提供酰基,它们的反应活性依次降低[17]。生物催化的酰化反应多采用脂肪酶。脂肪酶在体内催化的反应是酯水解,但通过控制条件反应亦可向生成酯的方向进行。脂肪酶相较于其他酶类有较多优点:首先,脂肪酶种类繁多。众多商业化酶类如猪肝酯酶(pig liver esterase, PLE)、铜绿假单胞菌脂肪酶(lipase,PAL)、褶假丝酵母脂肪酶(lipase,CRL)、猪胰脂肪酶(porcine pancreatic lipase,PPL)、荧光假单胞菌脂肪酶(lipase,PFL)等都可以有效催化轴手性化合物的不对称反应;其次,脂肪酶的底物范围较广,从联烯到联芳基底物等经脂肪酶催化都可获得良好的转化率和立体选择性(用ee表示);最后,脂肪酶具备高度耐受性,在有机溶剂中也可以保持良好的活性和稳定性[18],对于工业应用有较大优势。
图1 PLE 催化rac-1 的动力学拆分[19]
Fig.1 Kinetic resolution of rac-1 catalyzed by PLE[19]
脂肪酶催化联烯手性拆分的首个案例发表于1986年,多伦多大学的Jones等采用PLE催化三取代或四取代的联烯羧酸甲酯-1水解得到具有一定手性纯度的产物()-2与()-1(图1)[19]。2002年,佩鲁贾大学的Cipiciani[20]从酶的处理、条件优化、量级反应等展开研究,利用异丙醇处理的CRL(PT-CRL)催化KR反应,获得0.4 g产物()-4,ee达到了98% (图2)。以上方法均采用了商业化酶,通过条件优化和传统的动力学拆分反应,获得了高手性选择性的联烯类轴手性产物。此类联烯烃经过环异构化反应后,均可获得五元杂环,在天然产物制备方面具有广泛的用途。使用商业化酶具有一定优势,如原料易得、易于进行反应的扩大化等,但是由于商业酶并非针对轴手性底物开发,其应用范围受到严重限制,因此进行酶的理性设计与改造成为提升反应效率的重要手段。
图2 CRL催化联烯酯的动力学拆分[20]
Fig.2 Kinetic resolution of allenic esters catalyzed by CRL[20]
为充分拓展轴手性底物的酰化反应范围并且提升手性选择性,斯德哥尔摩大学的Bäckvall利用组合活性位点饱和测试CASTing技术[21]对PAL进行了定向进化。仅从600个变体中筛选出催化联烯酯5效果最佳的Leu162Phe变体,产物()-6的ee达到96%,手性选择性值为111,是野生型菌的13倍(图3)[22]。通过理性计算得知由于联烯5的C=C双键和酶蛋白162位的苯丙氨酸相互作用形成π-π共轭效应,使针对联烯的酶活有效提升。此研究有效利用了半理性设计定向进化技术使针对轴手性联烯底物的酶催化手性选择性得到改善,为轴手性底物的酶的改造开拓了道路。
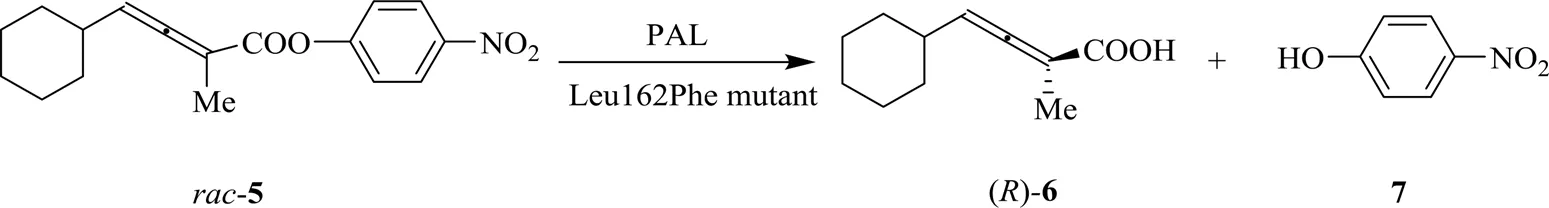
图3 PAL催化联烯酯水解[21]
由以上联烯酯水解反应的高手性选择性可知,其逆反应酯化反应也应该具备良好的选择性。斯德哥尔摩大学的Deska等针对联烯的酯化反应筛选了CRL、PPL、PFL以及洋葱假单胞菌脂肪酶(lipase,PSL)等数个脂肪酶[23],其中由PPL催化的轴手性联烯伯醇酯化反应的值高达200(图4)。此反应随后被拓展至11种联烯伯醇类底物。而手性联烯可以作为天然真菌代谢产物反应产物(-)-striatisporolide A的合成中间体。在酶催化的动力学拆分基础上,作者进行了(-)-striatisporolide A的全合成,产物的ee高达97%,转化率为67%。此研究良好地拓展了生物催化轴手性联烯反应的应用范围,并首次将生物催化反应应用于天然产物的全合成中,证明了此方法广阔的应用前景。

图4 PPL催化联烯酯水解制备天然产物[23]
除联烯类化合物外,联芳基化合物是最典型的轴手性化合物之一。联芳基化合物可作为合成药物分子、催化剂骨架、生物传感器表面材料等具有重要工业价值产物的手性砌块。其中,经典的具有光学活性的联萘酚(1-(2-hydroxynaphthalen-1-yl)naphthalen-2-ol,BINOL)是重要的手性催化剂和手性助剂。1985年,东京工业大学的Ikekawa首次利用微生物(sp.L-75、K-71)催化联萘双酯不对称拆分获得光学纯BINOL[24],开辟了此领域研究的先河。
2003年,布鲁克大学的Holland等[25]探讨了假单胞菌sp. 脂肪酶和PFL催化BINOL衍生物10的酰化反应(图5)。以BINOL为底物时,仅来自假单胞菌的脂肪酶具有活性,得到产物的ee值为96%;而以6,6'-二溴-BINOL 10为底物时,来自2种菌的脂肪酶均有活性,产物的ee值为93%~94%。2个酶催化的反应产物均为构型的单酯,没有双酯生成。此研究拓展了脂肪酶催化BINOL衍生物的底物范围,且衍生物10可通过钯催化剂催化进行Suzuki反应,具有一定的应用价值。
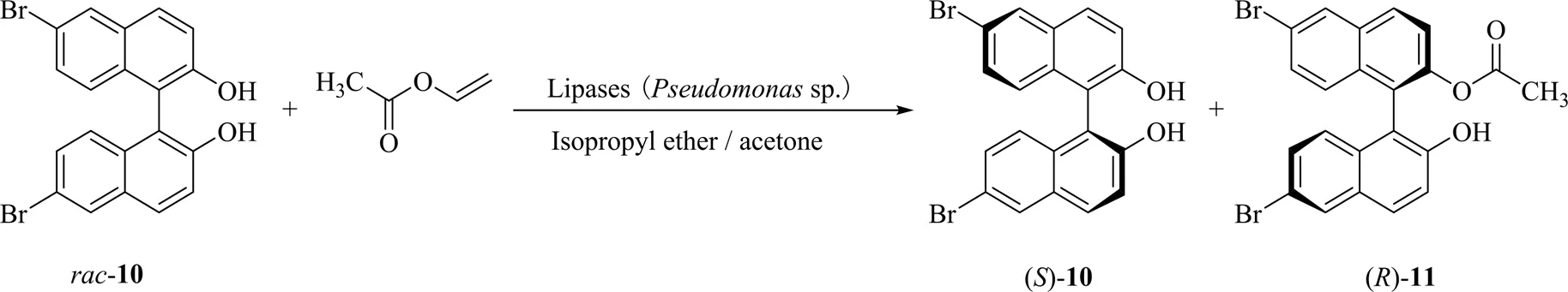
图5 来自于假单胞菌(Pseudomonas sp.)和荧光假单胞菌(Pseudomonas fluorescens)的脂肪酶催化BINOL衍生物的酯化反应[25]
除脂肪酶外,胆固醇酯化酶(bovine cholesterol esterase,CE)也可催化BINOL的喹啉类衍生物的水解反应。俄勒冈州立大学的Blakemore[26]等用CE成功催化了BINOL衍生物联喹啉双戊酸酯12的动力学拆分水解反应(图6(a)),ee值高达99%。但是底物2,2'-叔丁基取代的联喹啉双戊酸酯在CE催化下未能成功获得高手性纯度的产物,这可能是由双叔丁基取代导致的高位阻所致。作者通过利用手性纯的联喹啉双戊酸酯()-12与叔丁基锂反应获得对映体富集的2,2'-叔丁基取代的联喹啉双戊酸酯,随后进行水解,可获得ee为89%的手性产物()-14,成为获得高位阻的手性纯联喹啉衍生物的一种替代方法(图6(b))。此方法虽未直接利用生物催化获得手性纯的轴手性产物,但是当酶催化受到限制时,将生物方法与化学方法结合也不失为一种良好的替代方法。2016年,Chatterjee等[27]在链霉亲和素中引入生物素化的单膦钯复合物合成对映选择性的人工铃木酶,定点诱变优化这种人工金属酶的活性和选择性,生成的2-甲氧基-1,1'-联萘的ee最高可达90%。
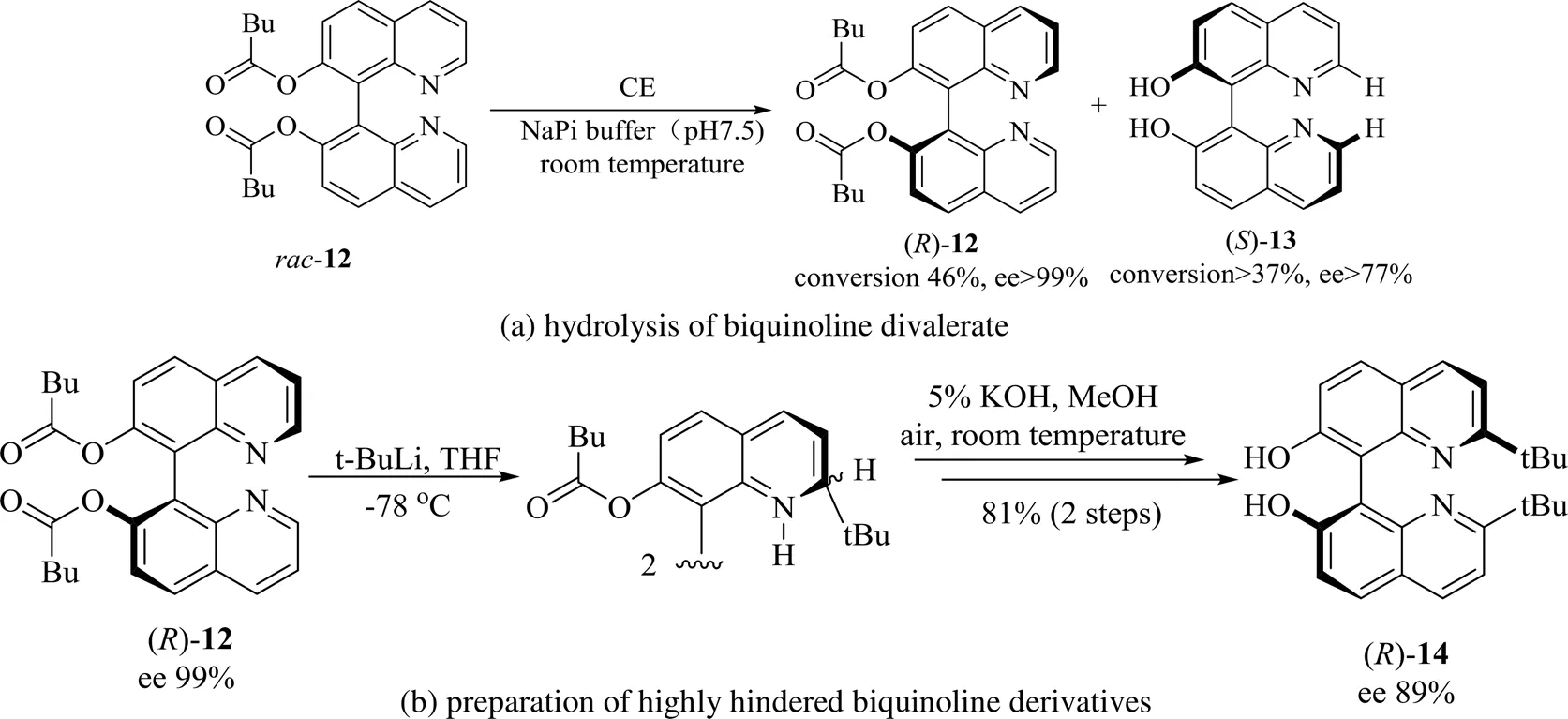
图6 CE催化联喹啉衍生物[26]
除酯化与水解反应以外,脂肪酶也可应用于酰胺化反应。山形大学的Aoyagi等[28]筛选了10种商用脂肪酶和多种酰基供体,探索了当联萘羧酸酯的烷基链长不同时脂肪酶选择性的改变。最后发现酰基供体的体积、烷基链长和脂肪酶的种类都对反应的手性选择性产生重要影响。得到当使用PAL、提升烷基链长以及增大酰基供体体积时,可使反应手性选择性提升的结论。而当联萘和羧酸酯之间的烷基链小于2个亚甲基时,无酰化反应发生;当间隔为2个亚甲基时,脂肪酶novozyme 435和chirazyme L-2可高效催化酰化的动力学拆分[29]。反应后未被酰化的底物()-15的ee值可达98%~99%(图7)。此研究首次为联芳基底物引入烷基侧链,证明了链长对脂肪酶催化活性的重要性,拓展了联芳基类轴手性反应的应用范围。

图7 脂肪酶催化联萘羧酸乙酯的酰胺化[29]
通过使用来自假单胞菌的脂肪酶类(species lipases,如PAL和PS)和来自南极假丝酵母的脂肪酶类(lipases,CAL如novozyme 435 和chirazyme L-2),Aoyagi还实现了肟衍生物的动力学拆分酰化与去酰化反应,获得较高的手性选择性(图8)[30-31]。novozyme 435 和chirazyme L-2催化-(1,1'-联萘)-2-甲醛肟酰化的反应速率较高,但手性选择性较低(<2)。而酮肟18由于与甲基亚胺相连的苯基对酶活性位点的空间位阻较大,导致酰化反应无法进行。相反水解反应则可顺利进行,PAL催化酰肟水解的ee值高于98%,高达222。此系列研究将脂肪酶催化的底物范围拓展至肟类的联萘基衍生物,拓展了应用范围。特别是空间位阻较大的酰肟类联萘底物17,展示了脂肪酶强大的催化能力及对底物广泛的接受范围。

图8 脂肪酶催化高位阻酮肟类底物的水解反应[31]
除联萘类底物外,联苯类化合物是合成抗生素、抗菌药物以及HIV逆转录抑制剂等药物的原料,具有重要的研究价值。而固定化酶在成本以及应用潜力上均优于游离酶。意大利生物化学研究中心(ICB)的Sanfilippo比较了四种不同固定化方法制备的PSL催化的联苯酚类底物的动力学拆分,其中以陶瓷为载体的固定化PSL-C催化的单酯()-20的ee值高达90%,为75(图9)[32]。由于轴手性化合物的特性及第1个酰基造成的位阻效应,使第2个酰基无法反应因此无双酯生成。其手性选择性与反应速率均高于未固定化的酶以及以高岭土为载体的酶PSL-D。
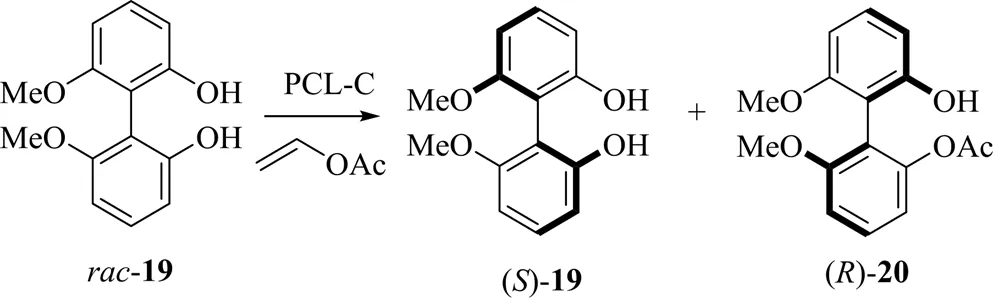
图9 PSL-C催化联苯酚的动力学拆分[32]
理论上,由生物催化构型不稳定的联芳内酯开环化可以获得手性纯的联芳基产物,然而Deska等[33]针对此反应测试了8种脂肪酶均未得到高手性纯度的产物,原因可能是羧酸酯附近的高位阻基团阻碍了反应的进行。但是高位阻基团却可以作为很好的亲核试剂参与转酯化反应,因此利用化学方法还原内酯得到联芳基伯醇21后,再与乙酸异丙烯酯反应可以获得手性纯度较高的转酯化反应产物22。通过筛选不同酶类发现米黑毛霉脂肪酶(lipase)催化可得ee值达86%,为7.8(图10)。

图10 脂肪酶催化联芳伯醇的动力学拆分[33]
脂肪酶在有机合成化学中的应用较其他酶类更为普遍,是生物催化剂中应用最广泛的一类酶。脂肪酶对有机溶剂高度的耐受性是它们具有如此重要地位的关键性原因。从水解反应、酯化反应、转酯化反应到酰化反应如表1所示,表中、分别为酶优先与底物的构型或构型。脂肪酶在重要工业化进程中起着不可或缺的作用。而由novozyme 435、PAL、PFL、PPL等成功的商业化酶催化的轴手性化合物的动力学拆分反应在生物催化领域占有举足轻重的地位。亟待进一步对轴手性底物的酶进行改造与开发,拓展底物应用范围,且将此类动力学拆分反应应用于天然产物与药物中间体的工业化合成中,为轴手性化合物合成的产业化奠定良好的基础。
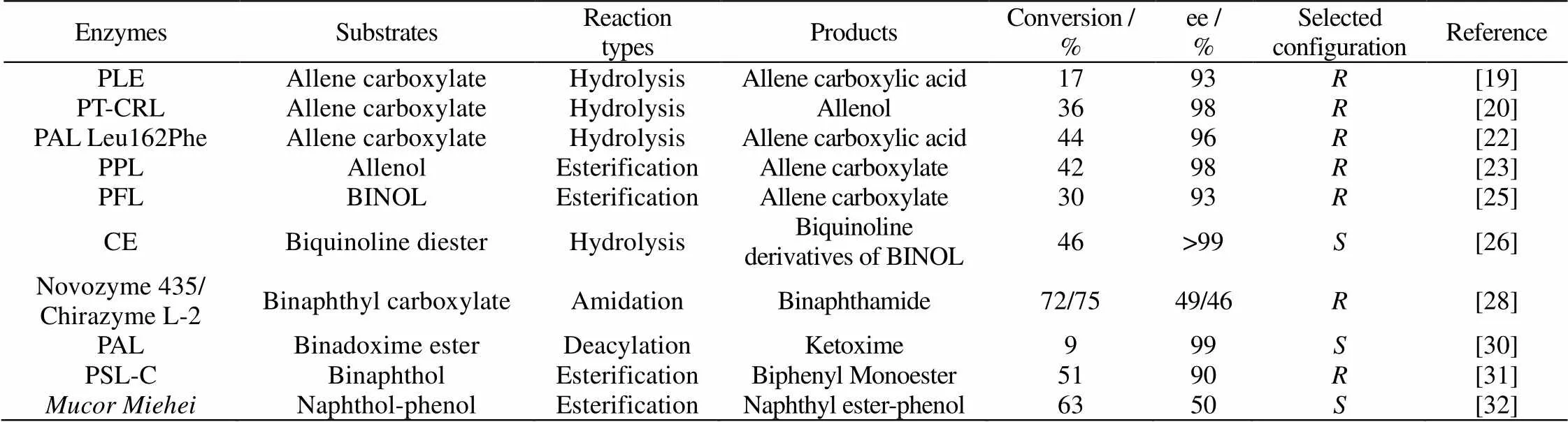
表1 本小节涉及的酰化反应汇总
2.2 氧化还原反应
除水解酶外,利用氧化还原酶催化不对称合成是获得高光学纯度的轴手性化合物的另一重要手段,近年来成为生物催化手性转化的重要方向。可应用于此类型反应的酶类有单加氧酶、双加氧酶、氧化酶和脱氢酶等[34]。尽管氧化还原酶具有高效的生物催化性能和高度立体选择性,但其专一性使底物范围狭窄、辅酶依赖严重、催化条件苛刻从而导致催化应用受限。高效氧化还原反应的实现需要注意酶的选择、反应方式及途径、反应介质、辅酶再生等因素[35]。探讨并解决这些问题对研究轴手性化合物的生物合成具有重要意义。
生物催化轴手性化合物的氧化还原反应的早期案例起于1980~1986年,马塞大学的Gil等探索了绿脓杆菌(ATCC17504)及来自酿酒酵母的醇脱氢酶YADH等催化α-联烯醇的氧化反应[36-38]。随后,1988年日本东北大学的Kawahara[39]课题组利用微生物面包酵母催化2-醛基-1,1'-联萘23还原成相应的醇(图11)。其中应用了全细胞作为生物催化剂,未添加辅酶及其循环再生系统,操作较为简单方便。产物()-24a的ee最高可达70%。
*indicates that 23b and 24b are inconsistent with the other configuration
针对氧化还原反应的KR研究较少,而其后期多应用于DKR及去对称化等反应。与酰化反应相比,氧化还原反应发展较为滞后,目前仍处于起步阶段。主要的限制性因素是氧化还原酶类对于辅酶因子的依赖导致经济性不足、对轴手性底物较大的空间位阻较为敏感以及底物范围狭窄导致的应用不广泛等。更多的轴手性底物氧化还原反应的应用可见2.2 与 3节DKR与去对称化的氧化还原反应。
2.3 小结
KR反应是最早应用于高手性纯度的轴手性底物的合成的反应类型,也是应用范围最广泛的反应之一。这得益于:(1)脂肪酶的研发起步早、商业化程度高,而脂肪酶主要的应用领域就是轴手性底物的KR反应;(2)KR反应体系相对简单,研发难度相对较低,早期是手性合成领域的热点方法;(3)KR反应可应用的底物范围相对较广等多个方面的因素。然而,由于KR反应最高理论转化率仅为50%,这造成原料成本较高和资源的浪费;且酯化和酰化反应均需要后续步骤去除酯基或酰基供体,造成步骤的增加,导致经济性差。这多方面的原因促使人们研发新的反应体系如DKR或去对称化反应,从而克服KR反应的瓶颈,为工业化应用开辟新的方向。
3 生物催化轴手性化合物的动态动力学拆分(DKR)反应
DKR反应能够有效克服KR反应理论转化率仅为50% 的缺点,免去后续目标产物的纯化步骤,减少资源浪费,在应用中具有更高的经济性与实用性。但是由于需要结合消旋化反应与动力学拆分反应,使反应体系更复杂、系统优化难度更高。其中,消旋化反应和动力学拆分反应均可通过化学或生物方法完成。本章节主要针对应用生物催化的动力学拆分反应与消旋化反应相结合的研究成果,讨论DKR反应的优势与限制性因素,探讨本领域研究方向。
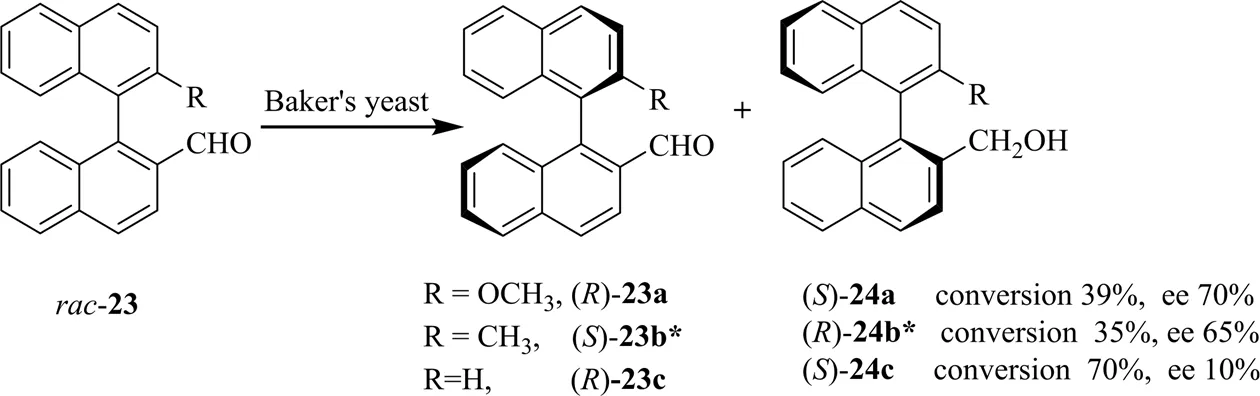
为拓展生物催化联烯类底物的应用,在Deska等以PPL催化11种联烯伯醇类底物的研究基础上[23],同时受到[Pd(OAc)2]/LiBr可催化联烯醇消旋化反应[40]的启发,Deska等[41]采用过渡金属催化剂与生物催化相结合的方法实现了联烯羧酸酯的DKR反应,如图12所示,图中n.d.表示ee值未定。研究采用与酶相容性较好的[(IPr)PdBr2]2催化剂进行联烯底物的消旋化,同时应用PPL进行选择性酯化反应。最终,产物转化率达70% 以上,(除26e外)ee均高于85%。此外,PPL可将86% ee的()-26a水解,获得91% 得率和ee>99%的()-25a。DKR反应克服了KR的缺陷,实现了化学与生物法的联用,极大地拓展了生物催化法的应用范围。
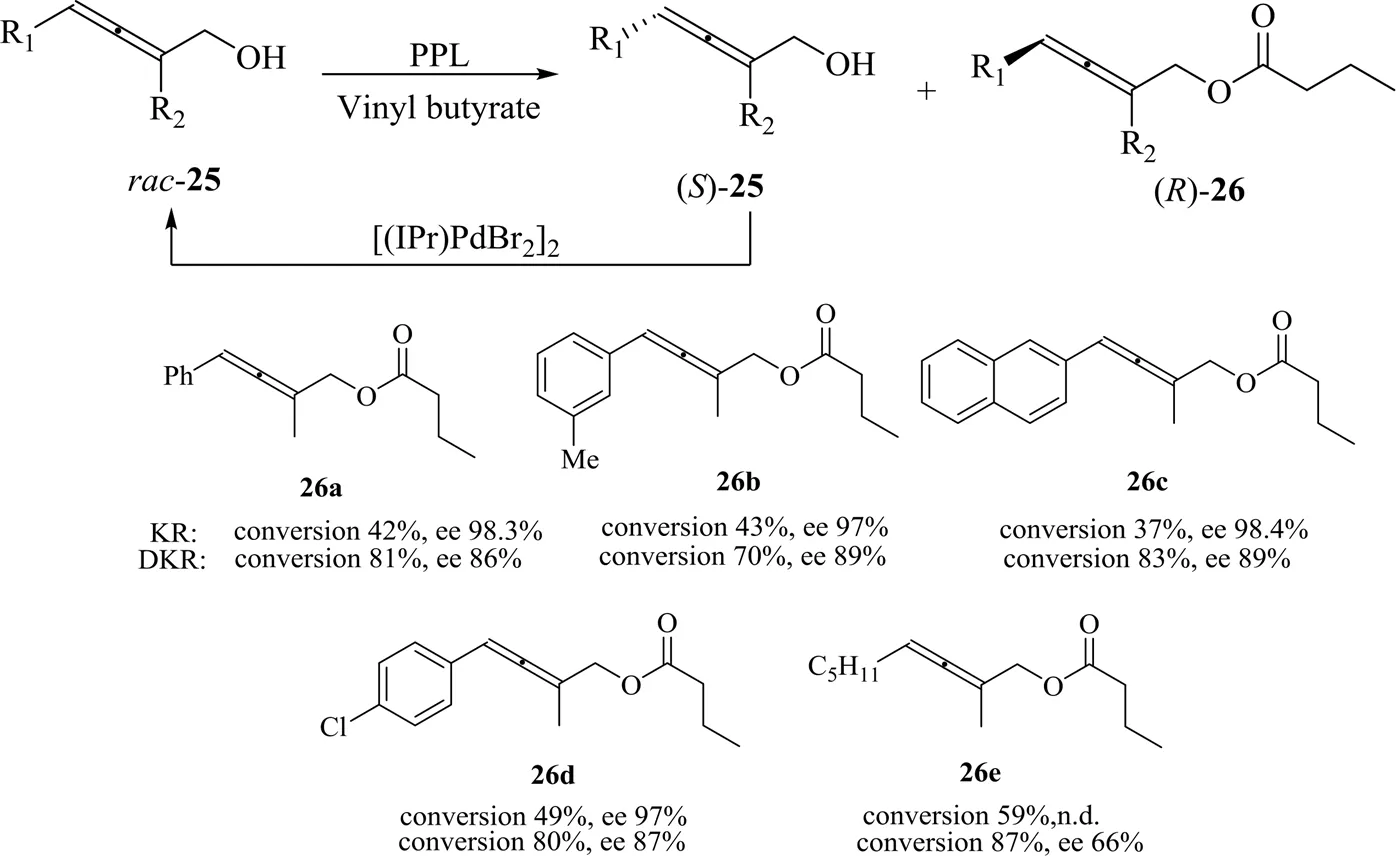
图12 PPL催化联烯伯醇酰化[41]
2005年,Sanfilippo等[42]利用PSL和Novozyme 435催化了硫醇类联苯底物27的DKR以及醇解反应(图13)。当试图利用PSL催化27的酯化时,硫醇并未被直接酯化,而是在生成了中间体28a和28b后,在酶催化下进行了酰化反应的动态动力学拆分,生成具有中心手性的29a和29b,经过进一步水解可获得具有轴手性的硫醇产物()-27,ee值高达91%。此方法虽然未能直接酯化硫醇而是通过间接的方式获得轴手性产物,增加了反应步骤,但是当直接酯化难以进行时,此方法以中心手性产物作为过渡性中间体最终获得高手性纯度的轴手性产物,创新性地解决了硫醇难以酯化的问题,为轴手性产物的制备提供了新的思路。
图13 PSL-D催化拆分硫代联苯[42]
除酰化反应DKR外,氧化还原反应也有少量案例报道。例如,酮还原酶(ketoreductase,KRED)被证明可以应用于位阻较大的联苯类中心手性底物的不对称还原反应[43]。KRED经过多轮定向进化和工业化生产,在医药中间体制备中应用非常广泛。曼彻斯特大学Turner课题组的Staniland等利用底物30和32的自然消旋化和KRED的还原反应相结合,完成了DKR反应,得到了高手性纯度的联芳基轴手性产物31和33(图14)[44]。为完成DKR反应,其KRED还原反应的反应速率必须低于底物消旋化的速率而高于产物消旋化的速率。所得产物ee值高于96% 且转化率高于83% (500 mg底物)。此反应巧妙利用了轴手性底物由于轴两侧位阻不同导致消旋化速率不同的特点,设计了两个轴不稳定的联芳基底物。这是首次关于联芳基类轴手性底物的DKR反应的报道,为氧化还原DKR反应的应用开拓了先河。

图14 酮还原酶KRED催化吡啶和异喹啉衍生物的动态动力学拆分[44]
综上所述,由于DKR克服了传统KR方法50% 理论转化率的限制,减少了原料的浪费,提高了生产过程的经济性,使此类反应成为轴手性化合物合成的最具前景的发展方向之一。但由于体系的复杂性,使DKR的研发周期增加,难度增大,也提高了研发的成本。这些难点成为DKR发展的限制性因素。随着酶的定向进化技术、辅酶再生循环系统、新消旋化与氧化还原反应的研发,我们期待未来有更多DKR反应系统性地应用于轴手性化合物的合成,扩展DKR反应在生物医用、食品与材料等领域的应用前景。
4 生物催化轴手性化合物的去对称化(desymmetrization)反应
除DKR外,去对称化亦可克服KR反应的最高理论转化率50% 的限制且无需引入化学催化剂。去对称化需要利用前轴手性的底物,通过酶催化的反应得到目标产物。如图15(a)所示,Deska等[45]以PPL催化联烯双醇34的去对称化,获得转化率为96%、ee为98% 的单酯()-35和少量过酰化的双酯36。将此反应应用于十个不同取代基的底物时,收率均高于90%,单酯ee为98%~99%。此反应应用范围甚广,例如可应用于胡椒基类天然产物Hyperione B的合成(图15(b))。经硝酸盐催化的环异构化反应可得产物ee高于99%。可见去对称化是合成高得率高对映选择性手性化合物的有效手段。

图15 PPL催化联烯双醇及其在天然产物中的应用[45]
为了进一步扩大底物范围,Deska还致力于四元取代的联烯双醇37的去对称化[18](图16)。相较于PPL,PFL表现出更好的催化效果:以较低的酶负载量可得反应转化率99%,单酯38a的ee高达97%。PFL催化不同取代基的底物去对称化,均获得了不错的转化率和ee(如表2所示)。
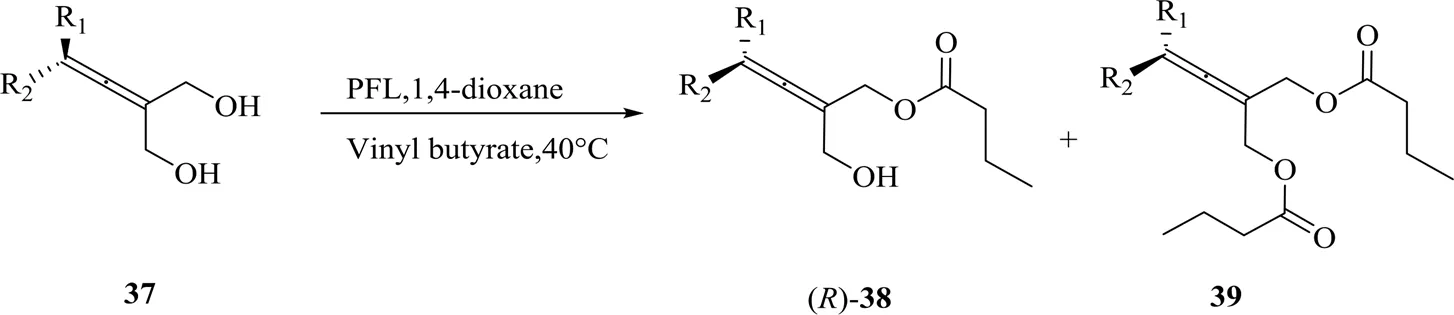
图16 PFL催化四元取代联烯双醇的去对称化[18]

表2 PFL催化四元联烯去对称化
除联烯外,联芳基轴手性底物也在手性配体、药物中间体合成等方面具有广泛用途。而目前生物催化此类底物的去对称化反应的研究仍较少。曼彻斯特大学的Yuan等[46]将半乳糖氧化酶GOase 的一个变体M3-5用于催化氧化前手性联芳基醚40[47],如图17所示,图中slow为反应速度较低fast为反应速率较高。GOase M3-5优先氧化40生成P构型的单醛41,随后选择性地氧化()- 41,通过进一步动力学拆分获得双醛42。这2步氧化反应都提升了()-41的手性纯度(ee约为93%)。这是首次将半乳糖氧化酶应用于轴手性化合物的合成方法,也是去对称化和动力学拆分相结合生物催化提高手性纯度的成功应用。此后,GOase M3-5的底物应用范围得到了拓展,联苯类底物43~48被氧化后都获得了手性纯度较高的产物[48]。这使半乳糖氧化酶成为轴手性底物氧化还原反应中应用最广的酶类。
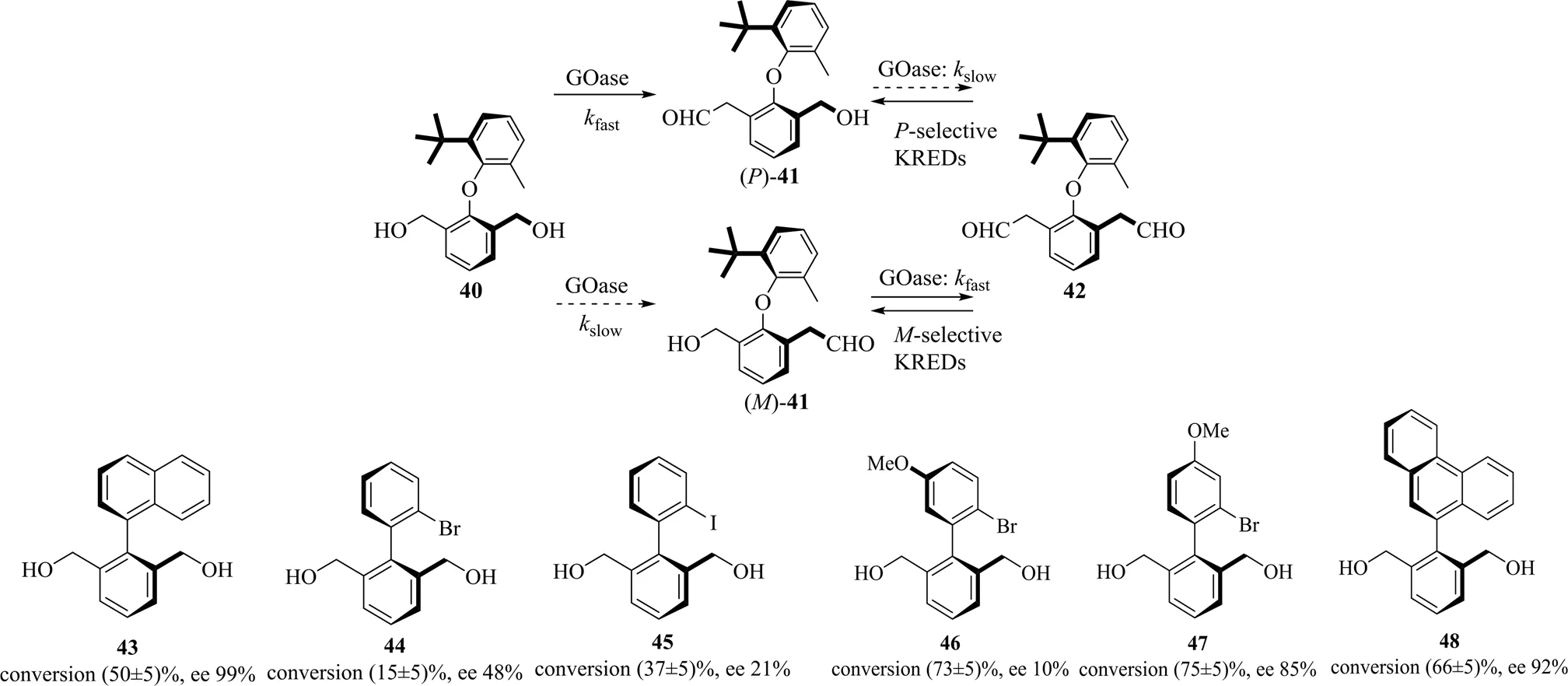
图17 半乳糖氧化酶催化联芳基化合物不对称合成[46]
除联烯和联苯基底物双醇的去对称化反应外,氧化偶联反应也可生成具有轴手性的产物。目前多数偶联反应均使用金属催化进行,使用生物方法进行的具有手性选择性的氧化偶联反应鲜有报道。一个典型案例是由过氧化物酶催化半棉酚自由基二聚生成棉酚,它是棉花中防御害虫和病原体的一个次级代谢产物[49]。南开大学汪清民课题组和霍恩海姆大学Schaller课题组[50]合作发现陆地棉中的dirigent蛋白GhDIR4起到了手性诱导作用,在漆酶催化氧化半棉酚49偶联为棉酚的过程中,()-(+)-棉酚50的ee值可达到80% 以上,而没有GhDIR4诱导的棉酚产物则是消旋的(图18(a))。这是天然产物中存在生物催化合成轴手性化合物的一个重要案例。类似的,弗莱堡大学的Müller等采用4种来源于不同真菌的漆酶催化萘并吡喃酮51(图18(b)),实现了不同程度的区域选择性和立体选择性的氧化二聚反应得到产物52[51]。

图18 漆酶催化氧化二聚反应[50]
除漆酶外,单加氧化酶P450也可以实现底物的区域选择性和立体选择性氧化偶联反应。Mazzaferro等[52]报道了7-去甲基铁苷53在黑曲霉菌属中P450酶DesC(在酿酒酵母中表达)的作用下,6和8'位偶联生成()-desertorin A 54;在细胞色素P450酶KntC[53]的作用下,8和8'位偶联生成55 ()-orlandin(图19)。这些偶联产物在O-甲基转移酶的作用下可以进一步生成半O-甲基化和完全O-甲基化合物:demethylkotanin、kotanin、desertorin B和desertorin C[54]。该研究不仅为全细胞生物催化剂催化生成新天然轴手性化合物方向提供了借鉴意义,也为黑曲霉作为潜在的生物合成工具铺平了道路。大阪大学的Haruna Nagayoshi等[55]评估了13种P450酶对 2,2',3,5',6-五氯联苯(PCB 95)和2,2',3,4,4',5',6-七氯联苯(PCB 183)的对映选择性氧化。这一研究为降解或消除有机污染物多氯联苯开辟了新思路。

图19 P450酶催化7-去甲基铁苷的偶联反应
这几个具有代表性的案例体现了氧化酶在天然产物和合成轴手性化合物上的良好表现以及生物催化剂诱导C─C耦合的巨大优势。生物催化剂的开发更是为环境污染提出新的解决方案,展示出广阔的应用前景。
与联萘双酯不同,萘-苯双酯、联苯双酯可以作为脂肪酶的有效底物。因此联芳基化合物也可以实现生物催化的去对称化。东京工业大学的Matsumoto等[56]进行了CAL和PSL催化联苯二乙酸酯的水解。并且将实验扩展到了1 g底物的规模,均得到了不错的转化率和手性选择性(图20)。但脂肪酶对萘-苯双酯和联萘双酯的选择性不同。
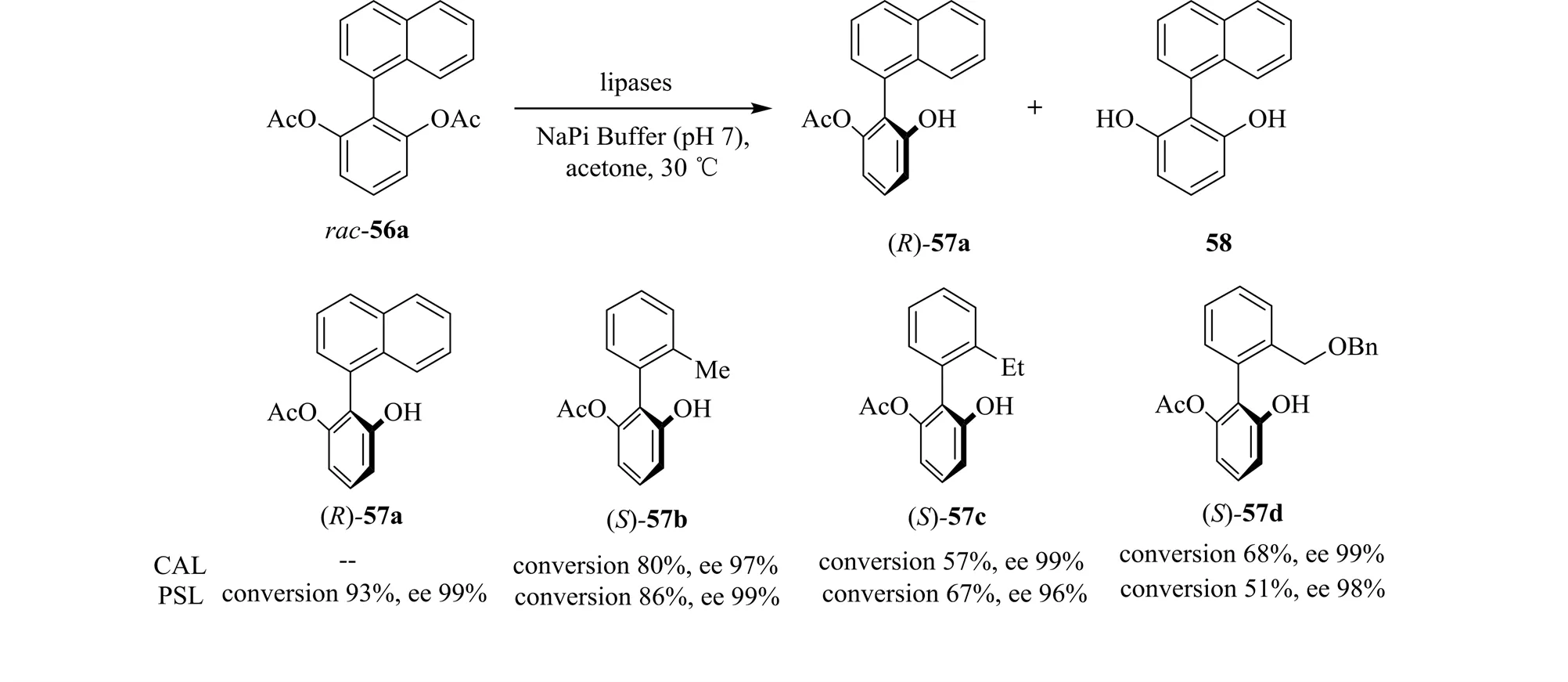
图20 脂肪酶催化联苯化合物的不对称转化
综上所述,去对称化反应由于反应体系仅需一种生物催化剂即可完成反应,相对于DKR方法较易于优化,同时也克服了传统KR方法50% 理论转化率的限制,因此近年来研究较为深入。但是去对称化反应需要对应的前手性体作为底物,因此反应设计和成本控制方面有更多工作尚待完成。其中,氧化还原酶参与的去对称化反应是近年来的研究热点。除DKR与去对称化反应外,针对轴手性底物的去消旋化反应(deracemisation)尚未见报道,这也是轴手性类化合物的生物催化反应在目前领域的缺失。总之,生物催化轴手性底物的研究目前仍为前沿的研究领域,亟待拓展能应用于轴手性底物的酶促反应类型,研发具有产业化价值的高值产物,为本领域的发展开拓崭新的发展平台。
5 总结及展望
本文总结了轴手性化合物的生物催化合成方法,从KR、DKR、去对称化等手性拆分方法入手,着重阐述了水解酶(脂肪酶)和氧化还原酶类在轴手性化合物的生物合成方向的应用。
随着手性拆分技术的发展,大量的高纯度手性化合物在医药中间体制备、手性配体合成及功能性材料等领域得到了重要应用。生物催化法具有高度的立体选择性、区域选择性和高效的催化效率,且大量商业酶的生产与制造降低了生物催化剂的成本;固定化酶和固定化细胞技术克服了酶在有机溶剂中活性低的困难,反应后极易从反应体系分离,有利于大规模的工业化生产;基因工程和蛋白质工程提高酶稳定性,改进酶活性,拓展底物范围,解决底物范围窄的问题,这一系列重要进展使手性药物的工业合成进入了生物催化的新世纪。然而,在轴手性化合物的生物催化合成领域,研究起步较晚,研究难度大、周期长,致使本领域发展稍落后于化学催化轴手性化合物合成。主要原因有:(1)与中心手性化合物相比,轴手性化合物分子大,水溶性差,与除脂肪酶外大多数酶的天然反应环境不匹配。这使定向进化中的高通量筛选成为难点,因此目前除脂肪酶外极少有针对轴手性底物的酶改造研究。(2)轴手性化合物体积大且空间位阻高,使底物难以进入酶的活性位点反应,致使许多酶在应用于轴手性底物时难以得到高手性纯度的产物。(3)轴手性底物合成难度高,使底物库的拓展较为困难。多数轴手性底物需要数步反应合成,转化率低,条件苛刻,致使底物库数量难以进行量级上的拓展。(4)酶催化成本难以降低,与化学方法相比不具优势。(5)基于目前的研究,作者认为轴手性底物的生物催化反应在诸多领域有着巨大的应用前景,但目前仍缺少较为成熟的技术将该方法融入最终产品的工业化合成过程中,如何将生物催化方法投入最终的生产中依然是今后重要的研究方向。
随着人们对轴手性化合物认识的加强以及生物催化和酶工程领域的不断突破,相信生物催化轴手性底物合成领域将迎来快速发展的阶段,在各个应用领域的广度与深度也会不断拓展,未来将会进入崭新的发展平台。
[1] CLAYDEN J, MORAN W J, EDWARDS P J,. The challenge of atropisomerism in drug discovery [J]. Angewandte Chemie International Edition, 2009, 48(35): 6398-6401.
[2] BRINGMANN G, GULDER T, GULDER T A MAtroposelective total synthesis of axially chiral biaryl natural products [J]. Chemical Reviews, 2011, 111(2): 563-639.
[3] KOZLOWSKI M C, MORGAN B J, LINTON E C. Total synthesis of chiral biaryl natural products by asymmetric biaryl coupling [J]. Chemical Society Reviews, 2009, 38(11): 3193-3207.
[4] LAPLANTE S R, FADER L D, FANDRICK K RAssessing atropisomer axial chirality in drug discovery and development [J]. Journal of Medicinal Chemistry, 2011, 54(20): 7005-7022.
[5] ZHANG D W, LI M, CHEN C F. Recent advances in circularly polarized electroluminescence based on organic light-emitting diodes [J]. Chemical Society Reviews, 2020, 49(5): 1331-1343.
[6] MIYASHITA A, YASUDA A, TAKAYA H,. Synthesis of 2,2′-bis(diphenylphosphino)-l,l′-binaphthyl (BINAP), an atropisomeric chiral bis(triaryl) phosphine, and its use in the rhodium(I)-catalyzed asymmetric hydrogenation of α-(acylamino)acrylic acids [J]. Journal of American Chemical Society, 1980, 102(27): 7932-7934.
[7] HAACK K J, HASHIGUCHI S, FUJII A,The catalyst precursor, catalyst, and intermediate in the RuII-promoted asymmetric hydrogen transfer between alcohols and ketones [J]. Angewandte Chemie International Edition, 1997, 36(3): 285-288.
[8] KNOWLES W S, SABACKY M J, VINEYARD B D. L-Dopa process and intermediates: US, 4 005 127 [P]. 1977-01-25.
[9] EVANS D A, WOOD M R, TROTTER B W,Total syntheses of vancomycin and eremomycin aglycons [J]. Angewandte Chemie International Edition, 1998, 37(19): 2700-2704.
[10] NICOLAOU K C, TAKAYANAGI M, JAIN N F ,Total synthesis of vancomycin aglycon—part 3: Final stages [J]. Angewandte Chemie International Edition, 1998, 37(19): 2717-2719.
[11] WILLIAMS D H, BARDSLEY B. The vancomycin group of antibiotics and the fight against resistant bacteria [J]. Angewandte Chemie International Edition, 1999, 38(9): 1172-1193.
[12] BRINGMANN GMENCHE DFirst, atropo-enantioselective total synthesis of the axially chiral phenylanthraquinone natural products Knipholone and 6′-O-methylknipholone [J]. Angewandte Chemie International Edition, 2001, 40(9): 1687-1690.
[13] DAGNE E, STEGLICH W. Knipholone: A unique anthraquinone derivative from kniphofia foliosa [J]. Phytochemistry, 1984, 23(8): 1729-1731.
[14] BRINGMANN G, MUTANYATTA C J, KNAUER M,Knipholone and related 4-phenylanthraquinones: structurally, pharmacologically and biosynthetically remarkable natural products [J]. Natural Product Reports,2008, 25(4): 696-718.
[15] BRINGMANN GTASLER S, ENDRESS H,Murrastifoline-F: First total synthesis, atropo-enantiomer resolution, and stereoanalysis of an axially chiral N,C-coupled biaryl alkaloid [J]. Journal of the American Chemical Society, 2001, 123(12): 2703-2711.
[16] GUSTAFSON J L, LIM D, MILLER S J. Dynamic kinetic resolution of biaryl atropisomers via peptide-catalyzed asymmetric bromination [J]. Science, 2010, 328(5983): 1251-1255.
[17] 钱清华, 张萍. 药物合成技术 [M]. 北京: 化学工业出版社, 2008.
QIAN Q H, ZHANG P. Drug synthesis technology [M]. Beijing: Chemical Industry Press, 2008.
[18] HAMMEL M, DESKA J. Enantioselective synthesis of axially chiral tetrasubstituted allenes via lipase-catalyzed desymmetrization [J]. Synthesis, 2012, 44(24): 3789-3796.
[19] RAMASWAMY S, RAYMOND A H F, JONES J B. Enantiomerically selective pig liver esterase-catalysed hydrolyses of racemic allenic esters [J]. Journal of the Chemical Society, Chemical Communications, 1986(20): 1545-1546.
[20] CIPICAIANI A, BELLEZZA F. Primary allenic alcohols of high optical purity via lipase catalyzed resolution [J]. Journal of Molecular Catalysis B:Enzymatic, 2002, 17(6): 261-266.
[21] CARBALLEIRA J D, KRUMLINDE P, BOCOLA MDirected evolution and axial chirality: optimization of the enantioselectivity oflipase towards the kinetic resolution of a racemic allene [J]. Chemical Communications, 2007(19): 1913-1915.
[22] REETZ M T, MARCO B, CARBALLEIRA J B,. Expanding the range of substrate acceptance of enzymes: Combinatorial active-site saturation test [J]. Angewandte Chemie International Edition, 2005, 44(27): 4192-4196.
[23] DESKA J, BACKVALL J E. Enzymatic kinetic resolution of primary allenic alcohols. Application to the total synthesis and stereochemical assignment of striatisporolide A [J]. Organic Biomolecular Chemistry, 2009, 7(17): 3379-3381.
[24] FUJIMOTO Y, IWADATE H, IKEKAWA N. Preparation of optically active 2,2′-dihydroxy-1,1′-binaphthyl via microbial resolution of the corresponding racemic diester [J]. Journal of the Chemical Society Chemical Communications, 1985, 19(19): 1333-1334.
[25] JUAREZ H M, JOHNSON D V, HOLLAND H L,Lipase-catalyzed stereoselective resolution and desymmetrization of binaphthols [J]. Tetrahedron Asymmetry, 2003, 14(3): 289-291.
[26] BLAKEMORE P R, MILICEVIC S D, ZAKHAROV L N. Enzymatic resolution of 7,7′-dihydroxy-8,8′-biquinolyl dipentanoate and its conversion to 2,2′-di-tert-butyl-7,7′-dihydroxy-8,8'-biquinolyl [J]. Journal of Organic Chemistry, 2007, 72(24): 9368-9371.
[27] CHATTERJEE A, MALLIN H, KLEHR J,An enantioselective artificial Suzukiase based on the biotin–streptavidin technology [J]. The Royal Society of Chemistry, 2016, 7(1): 673-677.
[28] AOYAGI N, IZUMI T. Kinetic resolution of 1,1′-binaphthylamines via lipase-catalyzed amidation [J]. Tetrahedron Letters, 2002, 43(32): 5529-5531.
[29] AOYAGI N, KAWAUCHI S, IZUMI T. Effect of the alkyl chain length of 1,1′-binaphthyl esters in lipase-catalyzed amidation [J]. Tetrahedron Letters, 2003, 44(30): 5609-5612.
[30] AOYAGI N, OHWADA T, IZUMI T. Facile synthesis of chiral 2-formyl-1,1′-binaphthyl via lipase-catalyzed acylation and hydrolysis of 1,1′-binaphthyl oximes [J]. Tetrahedron Letters, 2003, 44(45): 8269-8272.
[31] AOYAGI N, KAWAUCHI S, IZUMI T. Different recognitions of (E)- and (Z)-1,1′-binaphthyl ketoximes using lipase-catalyzed reactions [J]. Tetrahedron Letters, 2004, 45(27): 5189-5192.
[32] SANFILIPPO CNICOLOSIG, DELOGU G,Access to optically active 2,2′-dihydroxy-6,6′-dimethoxy-1,1′-biphenyl by a simple biocatalytic procedure [J]. Tetrahedron: Asymmetry, 2003,14(21): 3267-3270.
[33] SKROBO B, DESKA J. On the lipase-catalyzed resolution of functionalized biaryls [J]. Tetrahedron: Asymmetry, 2013, 24(17): 1052-1056.
[34] 张玉彬. 生物催化的手性合成 [M]. 北京: 化学工业出版社, 2002.
ZHANG Y B. Biocatalytic chiral synthesis [M]. Beijing:Chemical Industry Press, 2002.
[35] 聂尧, 徐岩. 生物催化立体选择性氧化还原中存在问题及其发展策略 [J]. 生物加工过程, 2008, 6(2): 1-9.
NIE Y, XU Y. Biocatalytic systems for stereoselective oxidoreduction: Existing limitations and development strategies [J]. Chinese Journal of Bioprocess Engineering, 2008, 6(2): 1-9.
[36] FERRE E, GIL G, BERTRAND M,Microbial transformations: A stereoselective pathway to oxidize allenic alcohols [J]. Applied Microbiology Biotechnology, 1985(21): 258-266.
[37] BERTRAND M, FERRE E, GIL G,Microbial oxidation of alpha-allenic alcohols [J]. Tetrahedron Letters, 1980, 21(18): 1711-1714.
[38] FERRE E, GIL G, BARRE M,. Oxidation of-allenic alcohols by yeast alcohol dehydrogenase [J]. Enzyme and Microbial Technology, 1986, 8(5): 297-299.
[39] KAWAHARA M K, MATSUMOTO H, HASHIMOTO H,. Kinetic resolution of 2-formyl-1,1′-binaphthyls by bakers′ yeast reduction of the formyl function [J]. Chemistry Letters, 1989,20(5): 1163-1164.
[40] HORVATH A, BACKVALL J E. Mild and efficient palladium(II)-catalyzed racemization of allenes [J]. Chemical Communications, 2004, 35(35): 964-965.
[41] DESKA J, OCHOA C D, BACKVALL J E. Chemoenzymatic dynamic kinetic resolution of axially chiral allenes [J]. Chemistry–A European Journal, 2010, 16(15): 4447-4451.
[42] SANFILIPPO C, NICOLOSI G, DELOGU G,Synthesis and biocatalytic resolution of a new atropisomeric thiobiphenyl: (2,2′,6,6′-tetramethoxybiphenyl-3,3′-diyl)dimethanethiol [J]. Tetrahedron: Asymmetry, 2005, 16(6): 1079-1084.
[43] TRUPPO M D, POLLARD D, DEVINE P. Enzyme-catalyzed enantioselective diaryl ketone reductions [J]. Organic Letters, 2007, 9(2): 335-338.
[44] SAMANTHA S, ADAMS R W, MCDOUALL J J W,Biocatalytic dynamic kinetic resolution for the synthesis of atropisomeric biaryl N-oxide Lewis base catalysts [J]. Angewandte Chemie International Edition, 2016, 55(36): 10755-10759.
[45] MANZUNA S C, BACKVALL J E, DESKA J. Enantioselective enzymatic desymmetrization of prochiral allenic diols [J]. Angewandte Chemie International Edition, 2011, 50(41): 9731-9734.
[46] TURNER N J. Enantioselective oxidation of C-O and C-N bonds using oxidases [J]. Chemical Reviews, 2011, 111(7): 4073-4087.
[47] YUAN B, PAGE A, WORRALL C P,Biocatalytic desymmetrization of an atropisomer with both an enantioselective oxidase and ketoreductases [J]. Angewandte Chemie International Edition, 2010, 49(39): 7010-7013.
[48] STANILAND S, YUAN B, GIMENEZ A N,Enzymatic desymmetrising redox reactions for the asymmetric synthesis of biaryl atropisomers [J]. Chemistry – A European Journal, 2014, 20(41): 13084-13088.
[49] LIU J, STIPANOVIC R D, BELL A A,Stereoselective coupling of hemigossypol to form (+)-gossypol in moco cotton is mediated by a dirigent protein [J]. Phytochemistry, 2008, 69(18): 3038-3042.
[50] EFFENBERGER I, ZHANG B, LING L,. Dirigent proteins from cotton (sp.) for the atropselective synthesis of gossypol [J]. Angewandte Chemie International Edition, 2015, 54(49): 14660-14663.
[51] OBERMAIER S, THIELE W, FURTGES L,Enantioselective phenol coupling by laccases in the biosynthesis of fungal dimeric naphthopyrones [J]. Angewandte Chemie International Edition, 2019, 58(27): 9125-9128.
[52] MAZZAFERRO L S, HUTTEL W, FRIES A,Cytochrome P450-catalyzed regio- and stereoselective phenol coupling of fungal natural products [J]. Journal of American Chemical Society, 2015, 137(38): 12289-12295.
[53] GIROL G C, FISCH K M, HEINEKAMP T,Regio- and stereoselective oxidative phenol coupling in[J]. Angewandte Chemie International Edition, 2012, 51(39): 9788-9791.
[54] HUGENTOBLER K G, MULLER M.Towards semisynthetic natural compounds with a biaryl axis: Oxidative phenol coupling in[J]. Bioorganic & Medicinal Chemistry, 2018, 26(7): 1374-1377.
[55] NAGAYOSHI H, KAKIMOTO K, KONISHI Y,Determination of the human cytochrome P450 monooxygenase catalyzing the enantioselective oxidation of 2,2',3,5',6-pentachlorobiphenyl (PCB 95) and 2,2',3,4,4',5',6-heptachlorobiphenyl (PCB 183) [J]. Environmental Science and Pollution Research, 2018, 25(17): 16420-16426.
[56] MATSUMOTO T, KONEGAWA T, NAKAMURA T,Facile and highly enantioselective synthesis of axially chiral biaryls by enzymatic desymmetrization [J]. Synlett, 2002, 33(19): 122-124.
Progress in biocatalytic asymmetric synthesis of axially chiral compounds
SHANG Ya-li, YUAN Bo, FEI Qiang
(School of Chemical Engineering and Technology, Xi’an Jiaotong University, Xi’an 710049, China)
Axially chiral compounds are valuable chiral compounds used as core skeleton of natural products, pharmaceutical intermediates and chiral ligands. Compared to asymmetric couplings catalyzed by metal catalysts or other asymmetric chemical methods, biocatalytic methods are mild, environmental friendly and highly selective. With the fast development of enzyme engineering and directed evolution, biocatalytic asymmetric synthesis of axially chiral compounds has attracted increasing research interest. This article reviews major progress in the area of biocatalytic synthesis of axially chiral compounds starting from different synthetic methods kinetic resolution (KR), dynamic kinetic resolution (DKR) and desymmetrization, and discusses the prospects of applications and existing problems of this area.
biocatalysis; axially chiral compounds; asymmetric synthesis; kinetic resolution; dynamic kinetic resolution; desymmetrization
1003-9015(2021)05-0775-14
TQ 55
A
10.3969/j.issn.1003-9015.2021.05.003
2020-11-27;
2021-01-27。
国家自然科学基金(32171462);国家自然科学基金青年基金(21706205);国家博士后项目(2018M633518);陕西省自然科学基金(2018JQ2062)。
尚亚莉(1996-),女,山西运城人,西安交通大学硕士生。通信联系人:费强,E-mail:feiqiang@xjtu.edu.cn
