m6A甲基化修饰在胃癌中的研究进展*
2021-11-02贾丽娟张云强综述张百红岳红云审校
贾丽娟 张云强 综述 张百红 岳红云 审校
作者单位:①甘肃中医药大学第一临床医学院(兰州市730000);②中国人民解放军联勤保障部队第九四〇医院肿瘤科;③中国人民解放军联勤保障部队第九四〇医院眼科;④甘肃省人民医院普外二科
胃癌(gastric cancer,GC)是消化系统最常见的恶性肿瘤之一,在世界范围内具有较高的发病率和死亡率,目前在全球癌症死亡率中位居第3 位[1]。GC 早期症状不典型,通常在进展期被发现。目前,主要以手术治疗为主,近年来GC 手术的规范化为患者的治疗策略奠定了较好的基础。同时,化疗药物、靶向药物、免疫治疗药物的研发及应用为不同发展阶段的GC 患者治疗提供了更多的选择性。尽管如此,GC 患者的5年总生存率(overall survival,OS)仍较低[2]。GC 的发生发展是一个多步骤、多因素和多基因参与的极其复杂的过程,因此深入研究GC 的发病机制成为人类亟待解决的难题。
近年来,N6-甲基腺苷(N6-methyladenosine,m6A)RNA 表观遗传修饰成为肿瘤研究的热点。研究已证实m6A RNA 修饰异常与多种肿瘤的发生发展密切相关,其通过影响RNA 的转录、加工、翻译和代谢等过程影响肿瘤的发生发展[3]。因此,本文就 m6A RNA 甲基化修饰在GC 发生发展中的作用以及化疗耐药做出综述,旨在从表观转录组学层面深入了解GC 的发病机制,为探索GC 靶向药物治疗提供新策略。
1 M6A 甲基化的概念
m6A 甲基化是指发生在RNA 中腺嘌呤(A)碱基的第6 位氮原子上的甲基化修饰,其是真核生物中mRNA 上最为常见的RNA 表观遗传修饰,不仅调控编码RNAs,也修饰长链非编码RNA(long noncoding RNA,lncRNA),主要富集在 mRNA 终止子附近的 3'非编码区(3' untranslated region,3'UTR)[4]。m6A 甲基化修饰为一种动态可逆的调控修饰,mRNA 在m6A甲基转移酶(m6A methyltransferase)的催化下发生甲基化,并在去甲基化酶(m6A demethylase)的作用下去掉甲基,重新恢复为腺苷;且甲基化的 mRNA 能够被m6A 识别蛋白(m6A recognition protein) 选择性识别结合,从而调节mRNA 的可变性剪接、出核转运、翻译和降解,最终调控转录后基因的表达。目前,m6A甲基化的蛋白主要分为甲基转移酶、去甲基化转移酶和m6A 识别蛋白。
m6A 甲基转移酶也称“编码器”(writers),主要由甲基转移酶样 3(methyltransferase like 3,METTL3)、METTL14、METTL16)、Wilms' 肿瘤蛋白 1 相关蛋白(Wilms' tumor 1-associating protein,WTAP)、RNA 结合模体蛋白 15(RNA binding motif protein 15,RBM15)、病毒样m6A 甲基转移酶相关蛋白(virus like m6A methyltransferase associated protein,VIRMA,又称为KIAA1429)、锌指CCCH 型结构域蛋白13(zinc finger CCCH domain-containing protein 13,ZC3H13)、Casitas B 系淋巴瘤原癌基因转化序列样蛋白1(Casitas Blineage lymphoma-trans forming sequence-like protein 1,CBLL1)又称为HAKAI,均为核心组成的甲基转移酶复合体催化mRNA 上的碱基发生m6A 甲基化修饰。在催化 m6A 形成的过程中,METTL14 与 METTL3 形成异二聚体发挥催化作用,其中METTL3 具有催化活性的亚基,负责将甲基增加到腺嘌呤第6 位氮原子;METTL14 通过结合mRNA 并协助甲基定位,负责识别底物;WTAP 通过稳定METTL3 与METTL14 的相互作用发挥调节亚基的作用;KIAA1429 通过募集甲基转移酶核心成分METTL3/METTL14/WTAP,介导3′UTR 和终止密码子区域选择性m6A 修饰;RBM15 可作为衔接蛋白招募WTAP/METTL3 复合物,使其选择性地在特定的m6A 共识别位点进行甲基化;ZC3H13 调控WTAP 在RNA 上的募集并促进m6A 修饰,主要负责与RBM15 的连接,发挥支架作用协助ZC3H13/WTAP/KIAA1429(VIRMA)/HAKAI复合物进行核定位,确保m6A 沉积到靶向转录本上调控m6A 修饰水平的动态变化;METTL16 是新发现的具有催化活性的m6A 甲基转移酶,与METTL3、METTL14 同源,可以靶向结合U6 小核 RNA(U6 snRNA)引起U6 第43 位的腺嘌呤发生m6A 甲基化修饰,从而影响U6 snRNP对mRNA 前体的剪接。
m6A 去甲基酶称为“消码器”(erasers),能够“擦除”mRNA 上的m6A 甲基。m6A 去甲基化酶的成分主要包括脂肪质量与肥胖相关蛋白(fat mass and obesity associated protein,FTO)基因和烷基化修复同源物3/5(alkylation repair homolog 3,ALKBH3)、ALKBH5,保证了m6A 的动态可逆过程,可去除m6A 甲基化,从而通过影响 m6A 甲基化腺苷去甲基化调控mRNA 加工、输出和代谢过程。
m6A 结合蛋白称为“读码器”(readers),能够选择特异性识别mRNA 的m6A 甲基化位点,主要有 YTH结构域家族蛋白(YTH domain family 1/2/3,YTHDF1/2/3)和(YTH domain containing 1/2,YTHDC1/2)、胰岛素样生长因子2 mRNA 结合蛋白1/2/3(insulin-like growth factor 2 mRNA binding protein,IGF2BP1/2/3)、异质核核糖核蛋白(heterogeneous nuclear ribonucleoproteins,HnRNPs) [异质核核糖核蛋白C(HnRNPC) 、异质核核糖核蛋白G(HnRNPG)、RNA 结合蛋白异质核蛋白A2B1(HnRNPA2B1)] 及真核细胞起始因子3(eukaryotic initiation factor 3,eIF3)。这些蛋白能与m6A 位点结合,从而促进 mRNA 选择性剪接、降解、翻译、核输出及稳定性。
2 m6A 甲基转移酶
2.1 METTL3
METTL3 是甲基转移酶复合物的主要催化成分,其表达异常可改变 m6A mRNA 的命运,从而影响GC细胞的增殖、转移、侵袭和凋亡。Yue 等[5]研究发现,METTL3 表达水平升高与患者的不良预后呈正相关,同时发现METTL3 有助于上皮间质转化(epithelialmesenchymal transition,EMT)过程和体内的转移。进一步研究发现在GC 中含1 的锌指MYM 型(zinc finger MYM-type containing 1,ZMYM1)为METTL3的m6A 靶标。METTL3 通过依赖m6A 的方式调控修饰 ZMYM1 mRNA, 提升ZMYM1 的稳定性;ZMYM1 通过招募 CtBP/LSD1/COREST 复合物与E-钙黏蛋白(E-cadherin)启动子相互作用,介导抑制Ecadherin 的转录而促进EMT 程序和转移,从而影响GC 细胞的浸润和转移。另有研究[6]发现METTL3在GC 中发挥致癌作用,敲除METTL3 可抑制GC 细胞的增殖、迁移和侵袭。机制研究发现下调METTL3可诱导失活AKT 信号通路,主要通过降低AKT 磷酸化水平和下游效应因子p70S6K 和cyclin D1 的表达而抑制GC 细胞增殖;同时,METTL3下调增加了凋亡正性因子Bax 和caspase-3 表达,降低了凋亡负性因子Bcl-2 的表达,进而激活凋亡通路促进细胞凋亡。该研究提示下调METTL3 可能通过失活AKT 信号通路和促进细胞凋亡而抑制GC 细胞的迁移、转移和侵袭,有望成为治疗GC 的潜在靶点。Wang 等[7]的研究表明,METTL3 在GC 组织中的表达明显升高,并与不良预后密切相关。进一步发现p300 介导METTL3 启动子中H3K27 乙酰化活化促进METTL3转录,从而促进METTL3 的下游靶向基因HDGF 的表达,增加其mRNA m6A 修饰和提高HDGF mRNA的稳定性。分泌型HDGF 能促进肿瘤血管生成,而核型HDGF 能激活GLUT4 和ENO2 的表达增加细胞糖酵解的能力。该研究提示METTL3/HDGF/GLUT4/ENO2 轴通过增加糖酵解和血管生成的能力促进GC的发生和转移。Liu 等[8]研究发现,METTL3 在GC患者中显著表达,并且随着肿瘤分期和分级的进展,METTL3 的表达水平逐渐升高。此外,敲除METTL3可降低EMT 相关蛋白α-平滑肌肌动蛋白的表达,从而抑制GC 细胞增殖和迁移能力。另有研究[9]报道了METTL3 高表达与GC 患者的临床病理特征和较差的生存期显著相关,敲除METTL3 能有效抑制细胞增殖、迁移和侵袭能力。进一步RNA-seq 和m6Aseq 分析发现METTL3 可通过调控MYC 靶基因上的关键蛋白(如MCM5、MCM6 等)介导m6A 修饰而促进GC 发生发展。此外,Yang 等[10]进一步研究发现在GC 组织和细胞中METTL3 和MYC 的表达均上调;并且METTL3 通过提高MYC m6A 甲基化水平,促进了MYC 蛋白的翻译,从而增强GC 细胞的增殖、迁移和侵袭。Jiang 等[11]研究发现,在GC 细胞中敲除METTL3 可导致GC 细胞中细胞因子信号抑制(SOCS)蛋白家族SOCS2 表达增加,且SOCS2 的表达与GC 细胞增殖呈负相关。提示GC 细胞中METTL3 下调可诱导SOCS2 表达增加而抑制GC 细胞增殖。最近的一项研究[12]发现,m6A 和METTL3表达水平在GC 中升高,且METTL3 表达升高提示患者的恶性程度较高及预后较差。METTL3 通过依赖m6A/DGCR8 的方式促进pri-miR-17-92 进入致癌miR-17-92 簇,而miR-17-92 能够靶向抑制肿瘤抑制因子PTEN 或TMEM127 的表达激活肿瘤AKT/mTOR通路,从而促进GC 的生长和转移。
2.2 METTL14
METTL14 在GC 中发挥抑癌作用。Liu 等[13]通过分析临床样本和生物信息学方法研究发现METTL14 在GC 组织的表达水平低于癌旁组织,且METTL14 表达与TNM 分期显著相关;Kaplan-Meier 显示METTL14高表达患者的预后优于METTL14低表达患者。机制研究发现METTL14 下调增加了肿瘤激活因子PI3K、AKT 和mTOR 蛋白磷酸化水平,而METTL14 过表达降低了上述蛋白的表达而抑制GC 细胞增殖和侵袭。此外,研究METTL14 与EMT相关性,发现METTL14 过表达组中EMT 相关的蛋白vimentin、N-cadherin、MMP9 蛋白低表达,而Ecadherin 蛋白高表达;而METTL14 下调时,上述蛋白的表达相反,从而促进EMT 过程增强GC 细胞的迁移和侵袭。提示METTL14 通过抑制PI3K/AKT/mTOR 通路和调控EMT 通路来抑制GC 细胞的进展和侵袭,有望成为GC 中一个新兴的生物标志物。
2.3 WTAP
WTAP 作为甲基转移酶复合物辅助因子,在调控m6A 甲基化修饰过程中具有重要的作用。最新的两项研究证实WTAP 在GC 中扮演致癌基因的作用。一项研究利用组织芯片和癌症基因组图谱(the cancer genome atlas,TCGA)数据集检测WTAP 的表达,发现WTAP 在GC 中高表达,并且其高表达与预后不良密切相关,证实了WTAP 的表达是GC 生存的独立预测因子。机制研究发现WTAP 的表达与肿瘤免疫调节相关,WTAP 低表达可通过增加免疫调节核心基因的表达增强肿瘤免疫功能,从而抑制GC 的增殖并同时提高患者的生存预后;而WTAP 高表达可抑制肿瘤免疫功能而提升GC 的增殖能力[14]。这提示WTAP可通过调节肿瘤免疫功能影响GC 的发生发展。另一项研究[15]利用m6A 免疫沉淀测序分析(m6A immun oprecipitation sequencing analysis,MeRIP-Seq)分析证实WTAP 和m6A 在GC 组织和细胞中的表达均上调,且WTAP 的高表达与GC 患者预后不良密切相关。机制研究发现WTAP 与GC 的Warburg 效应(又称有氧糖酵解,是指癌症中葡萄糖代谢和能量供应异常)有关;利用体外功能实验表明,WTAP 过表达能够促进肿瘤的增殖和提高糖酵解能力;而体内异种移植实验表明,沉默WTAP 可抑制肿瘤的生长。进一步使用MeRIP-Seq 和MeRIP-qPCR 研究发现肿瘤代谢指标HK2 为WTAP 的靶标,WTAP 可与3'UTR m6A 位点结合进而增强HK2 mRNA 的稳定性,从而促进GC 细胞的发生发展。该研究提示WTAP 通过增强HK2 表达和参与调节Warburg 效应在GC 中发挥致癌作用,为GC 的治疗提供了新的途径和靶点。
2.4 KIAA1429(VIRMA)
KIAA1429 作为支架连接m6A 甲基转移酶复合物的催化核心成分,其在GC 中的作用逐渐被发现。最新的研究[16]采用实时定量聚合酶链反应和蛋白印迹法检测KIAA1429 在GC 组织和细胞系中的表达,结果发现KIAA1429 在GC 组织表达较癌旁组织表达高。功能实验发现上调KIAA1429 可促进GC 细胞增殖,而下调KIAA1429 可抑制GC 细胞增殖。通过mRNA 高通量测序和RNA 免疫沉淀法(RNA immunoprecipitation,RIP)分析发现,c-Jun是KIAA1429潜在的调控基因。值得关注的是,KIAA1429 能通过非依赖m6A 的方式调控c-Jun 的表达;此外,c-Jun 过表达可解除KIAA1429 基因下调引起的GC 细胞增殖抑制。该研究提示KIAA1429 在GC 中发挥致癌作用,可作为GC 潜在预后生物标志物和治疗靶点。
2.5 METTL16
METTL16 是一种新发现的m6A 甲基转移酶,主要甲基化 mRNA 的 3'非翻译区的 m6A 位点。Wang等[17]研究发现,METTL16 在GC 细胞和组织中高表达,并且其高表达与GC 患者预后不良相关。体内外实验均证实METTL16 可促进GC 细胞增殖和肿瘤生长。此外,下调METTL16 可通过阻断G1 期向S 期转化进程抑制细胞增殖;进一步机制研究发现cyclin D1 是METTL16 的下游调控蛋白;敲除METTL16 可降低GC 细胞中m6A 的整体水平和cyclin D1 mRNA的稳定性及其表达。该研究表明,METTL16 在GC中具有促癌作用,通过介导m6A 甲基化水平增强cyclin D1 mRNA 的稳定性,上调cyclin D1 的表达,从而促进GC 细胞的增殖和侵袭,这为探索GC 治疗的有效策略提供了可能的途径。
2.6 RBM15/15B
RBM15 是Split Ends(SPEN)蛋白家族的成员,是保守的RNA 结合蛋白,其可以通过与剪接体成分相互作用与RNA 结合。研究者通过检索TCGA 数据库中GC 患者的生物信息学分析发现RBM15 与总生存率(overall survival,OS)密切相关;并且建立RBM15 的GC 预后风险预测模型,发现RBM15 的HR<1,可作为GC 的保护基因。进一步体外利用RTPCR 技术检测RBM15 结果显示,与正常胃黏膜细胞GES-1 相比,RBM15 在GC 细胞中弱表达且RBM15 mRNA 表达下调。此外,利用免疫组织化学检测GC组织和相邻组织中RBM15 蛋白水平,结果发现RBM15 在癌旁组织中高表达,在癌组织中弱表达[18]。该研究标明,RBM15 在GC 中可能发挥抑癌作用。然而,截至目前,关于RBM15 在GC 中的表达及功能的研究非常有限,未来仍有很大的发展空间。
3 m6A 去甲基转移酶
3.1 FTO
FTO 是最早发现的 m6A 去甲基化酶,研究发现FTO 与GC 的发生发展相关,有望成为监测GC 预后的重要分子靶标。研究者通过采用组织芯片免疫组织化学法检测GC 组织和癌旁正常组织中FTO 的表达情况,发现FTO 在GC 组织中的表达明显高于癌旁正常组织;进一步发现FTO 表达水平与低分化、淋巴结转移密切相关,且与TNM 分期呈正相关;Kaplan-Meier 分析显示,高FTO 表达与GC 患者预后不良显著相关[18-19]。Zhang 等[20]研究发现,低m6A 信号与GC的不良临床病理特征有关;机制研究发现FTO 过表达,可降低 RNA m6A 甲基化水平,激活 Wnt/PI3K-AKT通路而促进GC 的恶性表型。Yang 等[21]利用RT-qPCR和Western blot 检测结果显示GC 组织中MYC 表达升高;进一步研究发现,FTO 可以去除MYC 基因5′端m6A 修饰,降低GC 细胞中MYC m6A mRNA 表达水平,增加MYC mRNA 稳定性,进而提高MYC 在GC 中的表达,最终促进GC 的增殖;相反,沉默FTO后,MYC 表达下调,抑制GC 的增殖。该研究验证了FTO/m6A/MYC 轴在GC 靶向表观遗传修饰中的致癌潜力。
3.2 ALKBH5
ALKBH5 在GC 中异常表达,但目前其在GC 中的作用尚未明确。陆瑛等[22]研究发现ALKBH5 在弥漫性胃腺癌中的表达低于正常胃黏膜组织。进一步研究发现,ALKBH5 表达下调后,mRNA去甲基化能力下降,E-cadherin mRNA 的稳定性减低,从而使细胞的极性和骨架结构变得更为疏松而更具侵袭性。机制研究发现ALKBH5 过表达,可下调基质金属蛋白酶MMP-2、MMP-9 表达而降低EMT 水平,从而抑制GC 细胞转移和侵袭能力。然而,Zhang 等[23]的研究与上述的研究结果相反,该研究团队发现ALKBH5能够潜在结合lncRNA NEAT1(nuclear paraspeckle assembly transcript 1),且发现NEAT1 和ALKBH5在GC 中显著过表达,同时两者高表达与GC 中的侵袭和转移相关。进一步的实验证实,ALKBH5与NEAT1的表达呈正相关,敲除ALKBH5上调NEAT1 RNA 的m6A 水平,可抑制GC 中NEAT1 的表达;此外,敲除NEAT1 能够显著抑制GC 细胞的侵袭和转移。机制研究发现,ALKBH5 和NEAT1 通过正反馈调节EZH2(多梳抑制复合体的一个亚基)的表达加速恶性表型的形成,从而影响GC 细胞的侵袭和转移。该研究表明,ALKBH5 通过去甲基化lncRNA NEAT1促进GC 侵袭和转移,ALKBH5/ lncRNA NEAT1/ EZH2轴可能是GC 潜在的治疗靶点。
4 m6A 结合蛋白
4.1 YTH 结构域家族
YTH 结构域家族蛋白与含有m6A 的 mRNA 结合,可调控mRNA 的定位和稳定性,该家族蛋白与GC 的发生发展相关。一项研究[24]通过分析不同人类癌症数据库的多种生物信息,发现约7%的GC 患者发生了YTHDF1 突变,且YTHDF1 高表达与肿瘤高增殖侵袭率和总生存期较差相关。体内外实验均证实,下调YTHDF1 可抑制GC 细胞的增殖。机制研究发现YTHDF1 以依赖m6A 的方式促进Wnt 通路关键受体蛋白7(frizzled 7,FZD7)的翻译,从而增强FZD7 的表达,最终激活Wnt/β-catenin 信号通路促进GC 的发生。该研究结果证实了YTHDF1 及其m6A介导的Wnt/β-catenin 信号通路在GC 中的致癌作用。此外,另有[25]研究通过对TCGA 数据库GC 患者的m6A 基因进行全面分析,也发现YTHDF1 在GC 中的表达上调,且 YTHDF1的高表达与高危型GC 患者显著相关,提示YTHDF1 可能是GC 早期诊断的潜在靶点。最新的一项研究进一步证明YTHDF1 在GC 组织中高表达,且YTHDF1 的上调与GC 分期、癌灶、肿瘤大小及预后不良显著相关,是GC 患者生存不良的独立预后因素。体内外实验发现,下调YTHDF1 可抑制GC 细胞增殖和侵袭,以及抑制体内肿瘤的发生和肺转移,并可同时诱导细胞凋亡。RNAseq、MeRIPseq 和RIP-seq 的交叉实验表明,USP14 是YTHDF1的下游靶点,且USP14 表达上调与YTHDF1 表达呈正相关。进一步发现YTHDF1 能够以依赖m6A 的方式促进USP14 蛋白的翻译,从而促进GC 的发生和转移[26]。该研究表明,YTHDF1-USP14 在GC 的发生发展中具有重要意义,为GC 的预后、治疗或诊断提供了重要依据。Shen 等[27]分析公共数据库GC 患者中YTHDF2 的表达水平,发现YTHDF2 在GC 组织表达明显低于正常胃黏膜组织,且YTHDF2 的低表达与GC 临床分期和患者的生存预后密切相关;进一步通过RNA-Seq 实验发现YTHDF2 与叉头蛋白C2(forkhead box protein C2,FOXC2) 的表达呈负相关,且YTHDF2 可通过负向调控FOXC2 表达水平抑制GC 的恶性增殖。该研究提示YTHDF2 在GC 中扮演抑癌作用,有望为临床上治疗GC 提供一个潜在的靶点。
4.2 IGF2BPs
m6A 结合蛋白 IGF2BPs 在GC 中异常表达。以往的研究已经证实前蛋白易位因子SEC62 具有致癌功能,He 等[28]研究发现,SEC62 在GC 中呈高水平表达,且下调SEC62 可抑制GC 细胞的增殖同时促进细胞凋亡。机制研究发现,METTL3 与SEC62 mRNA相互作用可诱导修饰SEC62 mRNA 上的m6A,从而促进IGF2BP1 对SEC62 mRNA 的稳定作用,最终促进GC 的进展。Wang 等[29]研究发现,m6A 修饰基因IGF2BP1、IGF2BP2 和IGF2BP3 在GC 组织中的mRNA 表达显著升高。进一步研究发现,IGF2BP1 表达水平较高提示患者的总生存期较差,该研究表明IGF2BP1 在GC 中可能发挥致癌作用。有研究[30]为确定IGF2BP1 参与GC 细胞的增殖和转移情况,将IGF2BP1 siRNA 靶向转染到人GC 细胞SGC7901和MGC803 中,功能实验显示,下调IGF2BP1 能显著抑制GC 细胞的增殖。此外,敲除IGF2BP1 能诱导细胞凋亡并伴有Bax 表达升高和Bcl-2、Bcl-Xl 表达降低,同时导致GC 细胞周期S 期检查点蛋白质CDK2、cyclin A2 表达减少及细胞周期S 期阻滞,从而显著抑制GC 细胞的增殖和转移。免疫组织化学分析显示IGF2BP1 在GC 组织中高表达且主要在细胞质中表达。该研究结果进一步揭示出IGF2BP1 在GC 中发挥致癌作用。此外,Shen 等[31]研究发现,长链非编码RNA LINC01559 在GC 细胞中表达显著上调。功能实验检测显示沉默LINC01559 可抑制GC 细胞的增殖、迁移和EMT 过程。此外,染色质免疫沉淀(chromatin immunoprecipitation,ChIP)实验表明,作为转录因子的(zinc finger E-box binding homeobox 1,ZEB1)可与LINC01559 的启动子结合,促进GC 细胞中LINC01559 的表达;而LINC01559 通过招募IGF2BP2 稳定ZEB1 mRNA 的表达促进GC 的进展。该研究证实ZEB1 诱导的LINC01559 通过招募IGF2BP2 稳定ZEB1 mRNA 表达,从而加速GC 细胞增殖、迁移和EMT 过程。
4.3 EIF
EIF3 是最大的真核起始因子,具有m6A 的读码功能,在翻译起始进程中起着核心作用。He 等[32]研究发现, EIF3D 在GC 中表达上调,其高表达与GC不良临床预后显著相关。另有研究[33]使用cBioPortal和Oncomine 数据库发现在GC 样本中存在EIF3H基因改变(突变、缺失和扩增),并且 EIF3H mRNA 在GC 组织中也上调。机制研究发现下调EIF3H 可使细胞周期阻滞在G0/G1 期而抑制GC 细胞的增殖;同时通过上调促凋亡因子和下调抗凋亡因子诱导细胞凋亡。这些结果提示EIF3H 可作为临床治疗GC 的一个新的治疗靶点。此外,EIF3B 是EIF3 复合物的主要支架亚基,在mRNA 翻译、细胞生长和肿瘤发展中起着重要的调控作用。Ma 等[34]研究发现,EIF3B 在人慢性胃炎和GC 组织中表达上调,且EIF3B 的表达与GC的分期和进展有关。提示幽门螺杆菌(Helicobacter pylori,Hp)感染可上调GC 细胞中EIF3B 的表达,提示EIF3B 可能参与了Hp 的致癌过程。进一步发现下调EIF3B 可以通过调控癌相关基因的表达来抑制GC 细胞的增殖和转移。此外,另有研究[35]显示EIF3B 在GC 组织中表达显著上调,且EIF3B 高表达与肿瘤浸润深度、淋巴结转移、TNM 分期及不良预后显著相关。机制研究发现EIF3B 通过调控EMT 和STAT3 信号通路促进肿瘤细胞的迁移和侵袭;同时基因富集分析(GSEA)和Western blot 结果表明,上调EIF3B 表达能增强PI3K/AKT/mTOR 信号通路的活性,从而促进GC 的进展。敲除EIF3B 可以抑制移植瘤的生长和体内肺转移。该研究证实了EIF3B 在GC 发生发展中发挥着致癌作用,是GC 患者的独立预后因素,这可能有助于探索和发现GC 治疗的新靶点。
4.4 hnRNPs 超家族蛋白
hnRNPs 超家族蛋白中的部分蛋白被鉴定为m6A 甲基化结合蛋白质,它们的异常表达会影响mRNA 的稳定性、翻译、剪接以及肿瘤的增殖转移,但是目前其在GC 中的作用尚无大量的文献报道。Chen 等[36]研究通过检索TCGA 数据库分析GC 中hnRNPR 的表达,结果显示,与正常组织相比,HnRNPR在GC 组织中显著高表达。更重要的是,免疫组织化学染色结果显示:HnRNPR 在GC 组织中也显著高表达,且hnRNPR 的高表达与肿瘤的侵袭性密切相关。CCNB1 是一种有丝分裂特异性因子,其与CDK1 形成复合物调节细胞周期G2-M 的转变;CENPF 是着丝粒家族的成员,其负责调控肿瘤转移。机制研究发现hnRNPR 的表达与CCNB1、CENFP的表达呈正相关,hnRNPR 高表达可通过正反馈上调CCNB1 和CENPF 的表达,直接与CCNB1 和CENPF mRNA 相互作用调控GC 的细胞周期和肿瘤转移,从而促进GC 细胞的增殖与转移。该研究提示hnRNPR-CCNB1/CENPF轴可能是治疗GC 患者的潜在靶点[36]。提示hnRNPA2B1 在各类GC 细胞中均过表达,而在胃腺黏液癌中hnRNPA2B1 扩增最多,且hnRNPA2B1的过表达与低生存率相关。细胞功能实验发现hnRNPA2B1 通过促进细胞增殖和转移,同时抑制细胞凋亡来维持GC 的恶性表型。此外,机制研究发现BIRC5-202 是hnRNPA2B1 的关键下游靶点,hnRNPA2B1 可与几个核心剪接体共同表达,调控抗凋亡因子BIRC5 的可变剪接,增加致癌亚型BIRC5-202 的表达,从而促进GC 细胞增殖和转移[37]。
目前的研究结果表明,m6A 甲基化在GC 的发生发展中具有重要作用,未来可能会成为GC 治疗的潜在药物靶点,表1。
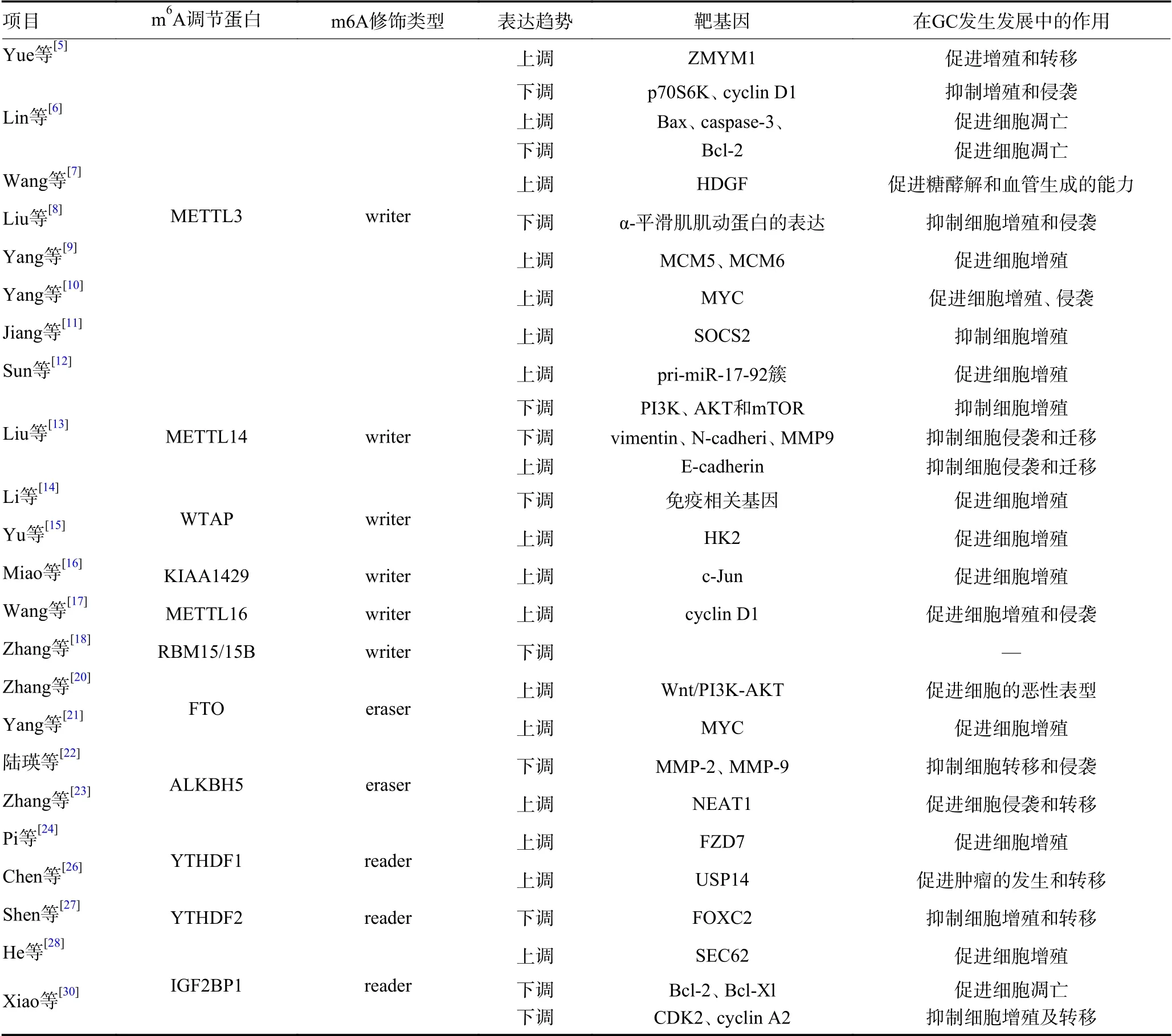
表1 GC 中m6A 调节蛋白的表达与功能
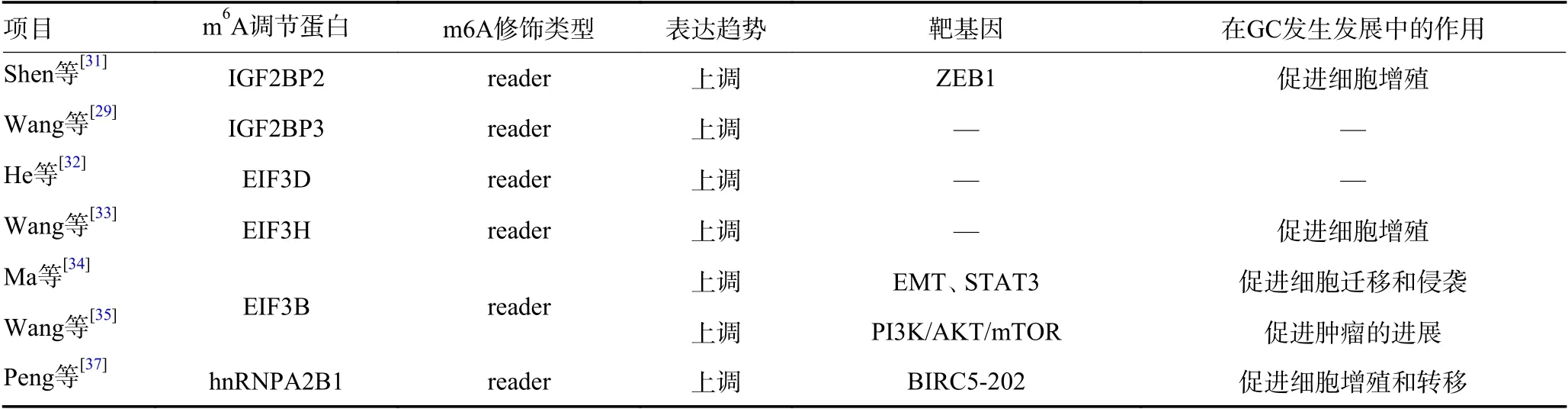
表1 GC 中m6A 调节蛋白的表达与功能(续表1)
5 m6A 与GC 化疗耐药
在肿瘤的治疗过程中,化学药物治疗有着不可替代的作用。但是,由于许多遗传和表观遗传信息的改变,化疗耐药仍然是恶性肿瘤治疗失败的最大原因。m6A 修饰相关蛋白在GC 化疗耐药中发挥重要的作用,有望成为GC 耐药治疗的新靶点。质子泵抑制剂可能是一种较好的治疗策略,可增加GC 细胞对抗肿瘤药物的敏感性。用奥美拉唑预处理可增强5-FU、DDP、TAX 对GC 细胞的抑制作用。Feng 等[38]研究发现,用奥美拉唑预处理可诱导FTO 表达下调而提升细胞总m6A 水平。进一步机制研究发现奥美拉唑诱导的FTO 抑制可增强激活mTORC1 信号通路,并抑制自噬,从而提高GC 细胞化疗药物诱导的细胞凋亡而提高疗效。同时,经奥美拉唑诱导的FTO 沉默通过依赖m6A 机制提高mTORC1 下游凋亡相关的肿瘤抑制基因DDIT3 的转录水平而促进细胞凋亡。该研究提示m6A 修饰FTO 可能在GC 中提高质子泵抑制剂奥美拉唑介导的化疗敏感性。此外,一项Ⅱ期研究中,证实了依维莫司对以前治疗过的晚期GC 患者显示出了良好的疗效。近期研究发现依维莫司可靶向干预GC 中METTL3/miR-17-92/TMEM127或PTEN/mTOR信号通路而提高化疗敏感性;进一步研究发现METTL3 高表达的GC 对mTOR 抑制剂依维莫司表现出更高的敏感性,并且依维莫司可以剂量依赖的方式逆转METTL3 诱导的肿瘤增殖。该研究结果揭示了METTL3 水平可能是依维莫司治疗GC 疗效的潜在预测因子,也为依维莫司治疗m6A/METTL3 水平较高的GC 提供了理论依据[12]。
6 结语与展望
综上所述,目前已证实m6A 甲基化修饰在GC 的发生发展中发挥重要作用,参与调控GC 的增殖、转移、凋亡和调节化疗耐药性,并影响着GC 的预后。但目前m6A 甲基化相关蛋白在GC 中的研究仍较少,亟需更多大规模的临床研究进一步阐明m6A 甲基化修饰调控GC 及诱发GC 的分子机制,探索可能作为GC 早期诊断和治疗的敏感性更高的m6A 甲基化标志因子,将有助于为临床GC 患者判断预后及探索新的靶向治疗创造更多的机遇。
