助催化剂负载调控氮化碳光催化制氢性能的研究进展
2021-10-25潘奕辰宜坚坚
潘奕辰,宜坚坚
(1.江苏大学 汽车与交通工程学院,江苏 镇江 212013;2.扬州大学 环境科学与工程学院,江苏 扬州 225127)
0 引 言
清洁能源的开发和利用已成了我国未来规划的重点内容和优先主题。基于我们目前所建立起来的能源储存和使用设施,氢能成为了代替化石燃料的最佳选择之一。目前主要的制氢方法是对烃类燃料(如甲烷)进行重整,虽然其产率高、技术成熟,但该反应需要消耗化石燃料,且不可避免的释放如二氧化碳(CO2)等气体,从可持续发展的角度来看并不够完美。近年来,光催化分解水制氢被认为是一种绿色、有前景的方法,然而其工业化仍然面临如效率低、反应设备放大困难等问题,而实现这一技术的基础是高效光催化剂的设计与制备[1]。近年来,各种有机、无机及钙钛矿材料均被发现具有光催化分解水产氢的性能[2]。
作为近年来新兴的有机半导体光催化剂,石墨相氮化碳(g-C3N4)具有不含金属元素、可见光响应、热稳定性和化学稳定性高,以及成分、形貌和化学结构可调节等优势[3]。得益于合适的能带位置(CB位置负于H+/H2,VB正于H2O/O2),g-C3N4在太阳能驱动的水分解理论上来说是可行的。然而,在实际的水分解反应中,g-C3N4粉末即使在牺牲剂的协助下也不能有效析出H2,其主要原因在于光生电荷从体相转移到表面的速率缓慢,以及其表面结构对H2O的吸附和活化能力不理想。因此,对g-C3N4进行合理的表面结构设计,从根本上解决上述缺陷从而促进其光催化产氢性能是十分必要的。表面助催化剂的负载是克服上述问题的一种可行策略,这主要是因为助催化剂的负载可以优化g-C3N4表面对H2O的吸附活化能力和其本身的电荷输运效率[4]。
在本综述中,作者将简要介绍助催化剂在促进光催化效率中的意义,对近年来通过在负载表面助催化剂提升g-C3N4产氢的研究案例进行合理的总结与讨论,重点关注助催化剂的种类、结构调控方法、负载量、尺寸效应等方面对调控g-C3N4光催化产氢性能的影响机制。本综述将为助催化剂/半导体型复合光催化剂的设计提供思路,并为调控光催化制氢的表面反应动力学速率的相关研究工作带来一定的参考。
1 光催化制氢的基本原理
自1972年学者发现以TiO2为光催化剂,可在紫外光照射下驱动水分解产生氢气的先驱性论文发表以来[5],光解水制氢研究方向引起了学界的广泛关注。发生该反应的过程如公式(1)所示。

(1)
由上述公式可知,分解水反应需要消耗的标准吉布斯自由能值为237.13 kJ·mol-1。热力学角度上不利于该爬坡反应的进行,需要额外的外部能量克服反应能垒。光催化分解水反应中,太阳能则成为了驱动水分解的外部能量。在此基础上,需要引入半导体光催化剂捕获光子能量来产生电子-空穴(e-h)对,驱动该反应的发生。为了驱动水分解反应的发生,半导体材料需要被高于自身带隙的光子能量激发才能产生电子-空穴对。半导体材料受光照激发产生的电子和空穴的氧化还原能力是由半导体的价带(VB)和导带(CB)位置决定的。当CB底部的电势比H+/H2的还原电位更负(0 V vs.NHE,pH =0)时,产氢半反应理论上才能进行。同样的,VB带边的电势需要较H2O/O2电势(1.23 V vs.NHE,pH =0)更正,产氧半反应才可以发生。因此理论上来说,用于分解水反应的半导体带隙应至少大于1.23 eV(不考虑过电位的理想条件),且跨越质子还原和水分子氧化反应的氧化还原电位[6]。
具体来说,光催化的水分解可分为吸收光子产生电荷、电荷分离和表面催化转化三个步骤,如图1所示。在光照条件下,具有合适能带结构的光催化剂激发价电子从VB端跃迁到CB端,同时在VB端留下了相应的空穴,形成电子-空穴对。随后,电子和空穴会从光催化剂的体相向表面(即催化反应发生的位置)转移。在此过程中,电子-空穴对的再次复合会限制能够到达表面催化位点的电荷数量,从而影响催化活性。最后,吸附在光催化剂表面的H+与表面汇集的电子发生氧化还原反应,形成氢气分子,并从光催化剂上脱附。在催化转化步骤中,理论上需要催化剂表面丰富和适宜的吸附和活性位点来促进反应的发生[7]。绝大多数光催化剂在不添加牺牲剂的情况下,纯水分解反应中只能生成微量的气体产物,导致机理研究难度较大。因此,为了探索催化剂结构参数与最终分解水性能之间的关系,目前很多反应仍需要加入牺牲剂来进行研究,因为理解产氢半反应是实现全分解水反应的基础。
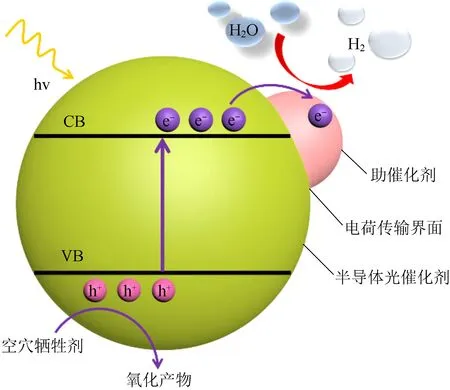
图1 光催化分解水制氢过程示意图Fig.1 Schematic diagram of photocatalytic production of hydrogen from water
2 表面助催化剂在光催化反应中的作用
g-C3N4具备合适的分解水制氢能带结构,理论上可以有效实现光催化制氢反应的发生。然而,g-C3N4的电荷迁移效率和表面H+吸附能不理想,严重限制了其高效分解水产氢。针对光催化剂的表面结构调整的方法众多,主要可以分为表面结构重筑、表面功能化和表面组装三大类[8]。表面结构重筑是不引入额外单元成分的情况下,对表面结构如表面积、成分、晶相、暴露晶面或缺陷等进行调整,使光催化剂的表面结构和物化性质更适合于光催化反应进行的一种策略;表面功能化是通过在光催化剂表面负载一些特定的功能化组分来优化单一光催化剂存在的一些固有缺陷的策略,如引入表面等离激元金属拓宽光谱吸收、负载助催化剂调控表面催化效率;表面组装策略表面来看与表面功能化存在类似,但表面功能化是负载不具有独立光催化性能的功能化材料,而表面组装则是由两种或者多种半导体构建的复合材料。对g-C3N4进行表面改性以促进光催化性能的方法中,负载助催化剂是提升光生载流子分离效率的有效方法,更重要的是,表面助催化剂还可以作为H+还原的活性位点,降低表面催化转化的过电位,以大幅提升分解水效率[9]。助催化剂除了能够促进基底半导体的活性外,还能抑制催化过程中可能发生的光腐蚀问题[10]。在g-C3N4/助催化剂体系中,g-C3N4作为吸光单元来产生光生电荷,而助催化剂的作用则是促进电荷动力学,将捕获的光生电荷在自身的活性位点上进行氧化还原反应。g-C3N4/助催化剂体系的合理构筑需要从能级结构、电子结构和界面结构等方面进行优化。只有这样,光生电荷才可以有效的从g-C3N4产生,以正确的方向穿过接触界面流向助催化剂表面。影响g-C3N4/助催化剂体系催化性能的因素很多,包括负载量、粒径、界面结合力等,都需要合理的进行考虑和设计[11]。
3 贵金属基助催化剂修饰氮化碳研究进展
具备大功函数、良好氢吸附能和稳定性的贵金属材料是负载在g-C3N4表面最常见的一类助催化剂。其中,Pt助催化剂在制氢反应中的效率非常理想,是目前公认的高效产氢材料。主要原因在于其具备有利于电荷迁移的大功函数、适中的氢吸附自由能(约-0.09 eV)以及优良的稳定性。g-C3N4表面负载Pt助催化剂通常可以获得几倍到几百倍不等的活性提升。Pt助催化剂的负载通常是通过还原含Pt前驱体(如H2PtCl6)实现,具体的方法包括原位光沉积、H2还原和NaBH4
还原等[12]。g-C3N4表面负载的Pt的形状和暴露晶面对催化活性存在显著的影响。如图2a-d所示,Cao等合成了具备不同形状和暴露晶面的Pt负载于g-C3N4的表面,为研究Pt的形状和暴露晶面对催化性能的影响提供了平台[13]。

图2 Pt负载的g-C3N4助催化剂Fig.2 Co-catalyst g-C3N4 loaded by platinum
研究数据显示样品的催化活性呈下列顺序:Pt纳米颗粒>Pt八面体>Pt立方体。结合理论计算和光电表征,作者分析认为Pt纳米颗粒可以优先暴露出更有利于质子吸附和还原的(111)晶面,从而得到最理想的效率。自2011年单原子催化报道以来,独特的高活性、高原子利用率等优势,使单原子在催化领域引领了一股潮流[14]。光催化反应中,经典的Pt/g-C3N4光催化剂在单原子催化领域也取得了一定研究成果。Li等人通过(图2e-g)g-C3N4特殊三嗪结构中N/C配位框架来锚定Pt单原子助催化剂,成功制备了单原子Pt/2D g-C3N4催化剂[15]。得益于Pt助催化剂原子利用率的最大化,所制备得到的单原子Pt/2D g-C3N4催化剂产氢反应中的转化频率(TOF)达到775 h-1,远高于Pt纳米颗粒和纳米团簇,证实了Pt助催化剂单原子化的优势,也说明了助催化剂尺寸的减小对于催化活性而言会存在促进作用。除了Pt以外,Rh、Pd等贵金属也被证实具备作为高效催化析氢助催化剂的能力[16-17]。虽然贵金属助催化剂的优势非常明显,但是其昂贵的成本,以及对于一些竞争反应(如分解水逆反应、氧还原反应)的高活性也带来了相应的局限性。
4 非贵金属基助催化剂修饰氮化碳研究进展
研究开发具有不含贵金属的高效助催化剂是目前g-C3N4基助催化剂研究的另一个热点方向。目前报道的可用作g-C3N4表面的产氢助催化剂众多,包括过渡金属、过渡金属氧化物/硫化物/氮化物/磷化物/氢氧化物、碳基材料、黑磷、氢化酶和人工分子等[11]。然而与贵金属助催化剂相比,非贵金属助催化剂的主要劣势在于低活性和低稳定性。因此,对助催化剂进行结构调控和性能优化是此方向的关键。以当前明星二维材料MoS2为例,其负载虽然可提高g-C3N4的析氢性能,但其效率受到导电性差和活性位点局限在边缘位置的限制。作为一种极具代表性的结构调控策略,Xu等对MoS2进行晶相结构设计,制备了1T-MoS2/O-g-C3N4二维异质结构,结合EXAFS和XPS对其晶相变化的精细结构进行了深入研究证实了MoS2具备八面体配位的1T相结构[18]。光催化产氢实验表明,作为助催化剂,1T-MoS2可显著提高单体O-g-C3N4的产氢性能,在低负载量(质量比重<0.2%)条件下甚至表现出优于Pt的活性。
该案例中,1T-MoS2通过原位溶剂热的方法负载在O-g-C3N4表面,有利于二者的紧密接触形成较强的界面相互作用,促进光生电子的传输效率。活性大幅增强的本质在于1T-MoS2优良的电导率有利于电荷迁移,以及相比2H-MoS2局限在边缘的催化位点,1T-MoS2具备丰富的基面和边缘催化位点(图3a)。
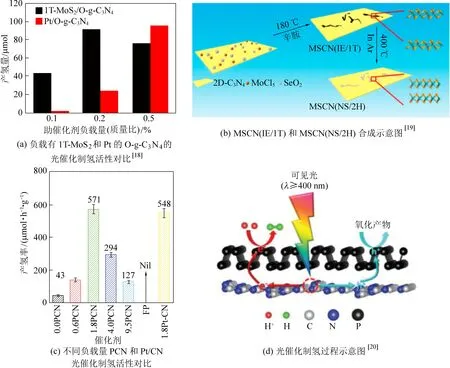
图3 非贵金属基助催化剂Fig.3 Non-noble metal-based co-catalyst
在晶相工程以外,Xu等在MoSe2助催化剂上报道了晶相和层间结构的协同调控(图3b)[19]。研究结果表明,在MoSe2的层间嵌入的正辛胺一方面可以撑开层间距,另一方面还可以作为电子供体,向Mo 3d轨道注入额外电子从而诱导2H-1T的相变。层间宽化1T-MoSe2和常规层间距2H-MoSe2在g-C3N4半导体表面负载后得到的复合催化剂表现出接近的光吸收和电荷分离能力,但产氢速率却显示出较大差距(1 672.6 μmol·h-1·g-1和186.7 μmol·h-1·g-1)。造成该现象的原因在于层间宽化及1T相结构有利于质子在MoSe2表面的吸附和催化转化。作为二维材料界的后起之秀,黑磷(BP)也被用作2D g-C3N4的助催化剂来促进其产氢性能(图3c-d)[20]。在18%乳酸水溶液中,BP/C3N4产氢率可达571 μmol·h-1g-1,高于同样条件下Pt/C3N4的产氢速率。需要指出,该数据是基于BP的最佳反应条件(包括负载量和牺牲剂的选择)得出的。较少量BP的负载会引起活性位点的密度不足,而过量的BP则会影响CN的吸光效率,都不利于催化活性的优化。BP/C3N4高活性的主要原因在于2D/2D界面上的紧密电子耦合、高电荷迁移率和BP表面丰富的产氢活性位点。在该工作中,BP的稳定性是仍待解决的一个弊端。图4通过比较外部量子效率(EQE)的方式,横向对比了g-C3N4表面负载不同类型助催化剂的性能差异。作者总结认为,尽管非贵金属基助催化剂的研究已经取得了长足的进展,但是和以Pt为代表的贵金属助催化剂相比,仍然存在差距。

注:CQDs/g-C3N4是405 nm测量,Ni/g-C3N4是440 nm测量,其他催化剂均为420 nm测量[18-19,21-28]图4 g-C3N4表面负载不同助催化剂的量子效率对比Fig.4 Comparison of quantum efficiencies of g-C3N4 loaded by different co-catalysts on the surface
5 结论与展望
总结来说,目前学者已经开发了一系列高效、稳定的助催化剂用于促进g-C3N4的分解水产氢活性。贵金属基的助催化剂具备理想的催化活性,其活性可通过调节尺寸、晶面、负载方法等手段进一步提升,但是较高的成本是其局限之一。非贵金属基的助催化剂本征活性较低,但是通过调控其晶相结构、强化与半导体间的相互作用等方法也可以将其活性有效提升,但是其活性和稳定性与贵金属材料相比仍然存在差距。在后续的研究工作中,作者认为以下三个方向可能会进一步促进该领域的发展,包括探索具备更高本征活性的新型催化材料、提升助催化剂在半导体基底上的分散性、利用精细表征与理论计算方法强化构效关系的理解。
