水产动物基因组近交分析软件的开发和应用
2021-10-18栾培贤张晓峰户国
栾培贤,张晓峰,户国
(中国水产科学研究院黑龙江水产研究所,黑龙江 哈尔滨 150070)
水产动物遗传资源是渔业生产和可持续发展的基础,也是满足未来不可预见需求的重要基因库。因此,世界主要国家均极为重视水产动物遗传资源的保护。在制定保护计划过程中,为使有限的人、财、物力发挥更好的效果,需要对保护的资源进行选择,区别需要保护的紧迫性和重要性,客观地评价物种及地理居群受威胁的状况[1]。受自然环境变迁及水利工程、过度捕捞、外来物种入侵等人类活动影响,我国东北、西北、西南等广大内陆地区自然水域以鱼类为代表的水产动物遗传资源都趋于衰退[2]。
一般说来,水产动物自然群体或小规模保种群体会发生小群体近交、个体生殖策略改变、个体小型化等情况,导致基因组某些位点的等位基因频率改变或者丢失,非等位基因组合发生显著变化[3]。这些基因组核苷酸水平的微小变化会体现在个体间亲缘系数改变,体质量、体型以及适应性等重要经济性状的加性效应和非加性效应方差组分变化[4]。有鉴于此,研发高效易行的水产动物基因组近交分析方法并在水生野生动物保护实践中应用,已成为保护遗传学和水产生物技术领域共同关心的重要问题之一。
本研究用高通量的SNP 分子标记替代传统家养群体中的系谱记录信息,拟开发精准快速的软件工具包,在无系谱信息鱼类的野生自然群体或捕获群体中构建实现分子亲缘关系矩阵,以多种分析方法解析并呈现群体遗传特征,提供如基因组共祖系数(Genomic coancestry coefficient)、显性亲缘关系(Dominance coefficient)、血缘同源(Identity By Descent,IBD)、状态同源(Identical By State,IBS)、基因组近交系数(Genomic inbreeding coefficient)、多维标度分析(Multidimensional Scaling,MDS)及群体遗传结构等统计基因组分析领域常见统计参数的估计值。本软件可以应用于水产动物遗传资源受威胁程度及保种群体规模需求量化分析方法研究,并为评价内陆珍稀水产动物小群体保种、亲本遗传管理及种群修复效果提供新的技术途径和科学可靠的参考依据。
1 软件的设计开发
1.1 软件设计流程
本软件以试验群体全部个体的全基因组范围内SNP 标记的分型数据为数据流起点;经过核心处理过程对输入数据进行全面的预处理,如样本分组、SNP 位点筛选、数据格式转换,将预处理后得到的最终数据集作为模型估计的输入信息;根据所框选的统计模型和分析算法完成基因组SNP 标记的各类亲缘关系参数的估计,最终以多种分析方法解析并呈现群体遗传特征,数据流和功能结构如图1所示。

图1 技术路线Fig.1 Technical route
1.2 软件结构设计
本软件采用数据处理层、模型工具层和可视化交互层的C/S 三层交互性构架结构设计(图2)。数据处理层包含SNP 基因分型数据集、个体和分组等数据的预处理;模型工具层包含集成的开源分析工具(内嵌了R、Perl 等软件及工作环境),可作为数据处理、模型建立、参数估计和计算结果统计分析的工作环境;可视化层包含工具包可视化交互环境,以及亲缘关系和群体遗传结构分析的可视化呈现。
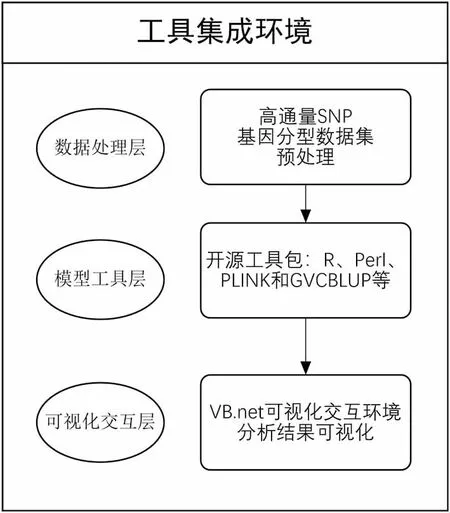
图2 软件开发的三层交互结构Fig.2 Three layers interactive structure in software development
1.3 开发工具与运行环境
本软件是在Windows 操作系统下,基于VB.NET 2012 语言、开源工具包(R、PLINK[5]和GVCBLUP[6]、LEA[7]、SnpSift[8])开发完成,可在安装有Microsoft.NET Framework 4.0 运行环境的Windows 10 操作系统上运行。为使本软运行稳定流畅,操作系统的硬件配置不低于酷睿i3 处理器、主频2.13 GHz、4 GB内存和128 GB 硬盘。
2 软件的功能实现
本软件通过原始数据提交、数据预处理、遗传分析等3 个功能模块实现其功能,具体情况如下:
2.1 原始数据提交模块
工具包使用者通过原始数据提交模块,选择需要分析的原始数据(VCF 格式文件)。原始数据提交后,工具包会自动解析原始数据信息,给出所提交数据集中样本数量和SNP 标记数量信息等相关信息,如图3 所示。
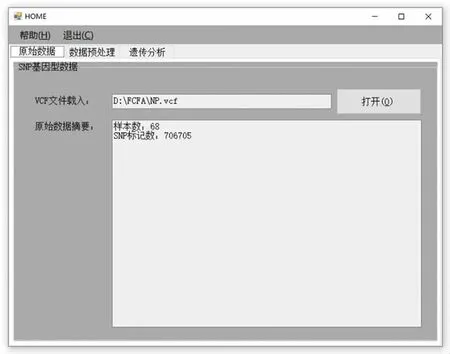
图3 原始数据提交模块Fig.3 VCF file submission module
2.2 数据预处理模块
该模块是数据分析前的重要预处理环节,用来对原始数据进行数据质量控制、SNP 标记筛选和样本标签的管理(图4)。选择有效的约束条件和控制标准,可以降低数据背景噪声,提高结果的准确性和可读性。使用者可以通过多种技术手段高通量获得全基因组范围内SNP 基因分型信息,如基因组重测序、简化基因组测序和基因芯片等。由于建库方式、测序量等不同,使全基因组范围内各个SNP 座位的数据完整性不同。该模块能够对原始数据进行质量控制,设定次要等位基因频率、哈温平衡检测显著水平、缺失基因型比例等阈值,使每个样本用于进一步分析的SNP 标记具有较好的一致性。

图4 数据预处理模块Fig.4 Data preprocessing module
基因分型文件内的样本标签多为测序分析公司内部编号。为了满足分析的实际要求,需将无意义的测序编号对应有序的一组个体编号上。此外,进行群体遗传结果分析时,也需要为个体所在群体指定群体编号。使用者需要将基因分型的个体编号与分析编号、群体编号相互对应,保存在文档中进行上传。
2.3 遗传分析模块
该模块是本软件的核心模块,集成了多种开源工具包,估计个体近交水平和个体间基因组亲缘关系,分析群体遗传结构等。使用者只需点选所需分析内容(图5),软件工具通过后台调用集成在本软件内的分析工具,即可在后台完成全部分析过程,输出相应结果。

图5 遗传分析模块Fig.5 Genetic analysis module
2.3.1 个体近交分析
该子模块基于全基因组范围内的SNP 标记估计个体近交水平,使用者通过个体分组信息,可进一步获取不同群体的近交水平等信息。该工具包提供两种个体基因组近交系数的估算方法:一是经典数量遗传学理论基于个体加性关系矩阵对角线元素计算个体近交系数;二是群体遗传学基于观测和期望纯合子关系评估个体近交系数。
2.3.2 亲缘关系分析
该子模块提供多种基因组亲缘关系参数估计方法,如共祖系数、显性亲缘关系、血缘同源(IBD)、状态同源(IBS)等,可从多个维度解析样本个体间的亲缘关系构成。使用者可以根据分析需求,选取关注的基因组亲缘关系参数,同时还能选择参数估计的模型。
2.3.3 群体遗传分析模块
该子模块基于高通量SNPs 标记计算分析常见群体遗传结构参数。多维标度分析(MDS)是一种类似于主成分分析(Principal components analysis,PCA)的降维分析方法。可在二维或三维空间展示个体间的基因组相似性,呈现出基于基因组相似性的个体空间散布图,展示群体结构。本软件还集成了LEA 工具,利用非连锁的SNP 标记估计群体遗传组成单元,推测现有群体中个体的原始来源。
3 实际应用案例
采用本软件,以利用SLAF-seq 技术分子标记开发的SNP 数据集为示例,按如下流程分析了新疆两个白斑狗鱼Esox lucius 群体共计68 尾个体(图6)。该数据集将酶切片段长度在414~464 的序列定义为SLAF 标签,预测到138 626 个SLAF 标签。实验中选择RsaI+HaeIII 的酶切方案,共得到189.71 Mreads,获得多态性的SLAF 标签313 663 个。

图6 野生鱼类亲缘关系和近交水平分析流程Fig.6 Analysis flowing chart of genomic relatedness and inbreeding of wild fishes
在数据预处理模块中,用户可以根据自身对测序数据集质量控制的需要设置相应参数,获得相应的数据集用于进一步遗传分析。本研究相关参数设置为测序质量值QUAL 不低于30,次要等位基因频率不低于0.05,位点哈温平衡显著水平设置为0.01,每个SNP 位点的完整性为95%,样本编号信息采用自定义编号。原始数据进行预处理之后,得到14 124 个位点的高质量数据。在遗传分析模块中,直接点选全部四种基因组亲缘关系分析方法,并勾选定义6 估计共祖系数和显性亲缘关系;选择PLINK 估计方法,估计个体的基因组近交系数。在群体遗传分析模块中,选择默认参数,用LEA 和MDS 两种分析方法显示群体遗传结构。
可以直接下载基因组近交系数估计值,也可将个体的近交系数估计值以柱形图的形式展示(图7)。
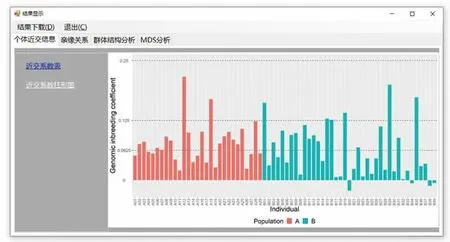
图7 近交系数柱形图Fig.7 Histogram of genomic inbreeding coefficient
共祖系数、显性亲缘关系、血缘同源、状态同源等基因组亲缘关系参数均以热图方式呈现。本文中以共祖系数为例,以热图形式展示相关结果(图8)。

图8 共祖系数热图Fig.8 Heatmap of genomic coancestry coefficient
基于LEA 推测群体构成结构(图9),用MDS方法展示个体空间散布图(图10),两种群体遗传结构分析的常用方法也可以直接以图形的方式从软件输出。

图9 个体遗传来源估计结果展示图Fig.9 Histogram of individual ancestral genetic source estimation
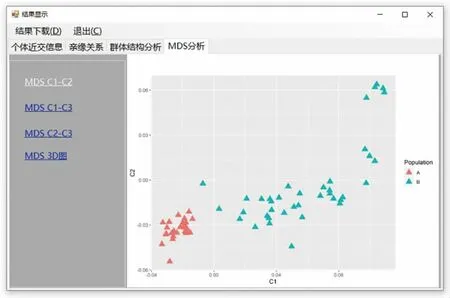
图10 二维MDS 散点图Fig.10 Two dimensional plot of MDS
4 分析与讨论
目前,增殖放流已成为水生生物资源养护的一项重要措施之一[9]。但是,野生自然群体没有遗传结构清晰的系谱记录信息,传统的标志放流方法无法准确评估有效群体规模、近交率等遗传灭绝风险指标,无法精确量化评价遗传资源濒危程度[10]。一些研究者和社会观察家认为,如果仅靠单纯数量意义上的增殖放流不仅会在经济、环境和增殖放流效果等方面不可持续,而且会降低野生群体的遗传多样性,使得种群遗传结构趋于简单化[11]。同样,野生自然群体没有遗传结构清晰的系谱记录信息,无法采用传统家养动物的遗传评定手段[12],有时甚至会出现放流种群非土著居群后代,占据了土著种群的生存空间和生态位,替代本地野生群体;或者放流群体与本地土著群体发生遗传交流,改变本地野生群体的遗传组成,破坏本地鱼类遗传资源。
现阶段,人们可以方便快捷并廉价地获得基于二代、三代测序技术的高通量SNP 分子遗传标记,应用这些标记信息就可以准确地估计野生水生动物的基因组近交水平。在开展渔业资源养护过程中,必须考虑水产动物基因组近交水平等因素。为此,本软件为一线野生水生动物保护工作者集成了现有依托于不同环境如R、Perl 及常用独立群体基因组学软件包的功能,提供了主流群体基因组和保护遗传学常用基因组参数估计,并利用VB.Net 予以界面可视化,实现全程鼠标点选即可实现高难度的群体基因组学分析。
遗传模拟分析中,本软件集成的各类方法理论上适用各种二倍体有性生殖动物。本研究还选取了新疆重要的土著经济鱼类白斑狗鱼群体的SLAF 简化基因组测序数据集作为本研究的实际案例,用于确认软件功能的有效性,也取得了实际效果。尽管如此,在样本量较大或受成本制约等情况下,有时仍会考虑使用SSR 等传统分子标记。因此,本研究也拓展了可以兼容SSR 标记的分子标记共祖分析方法[13,14],将在下一阶段软件升级时集成整合。该软件的推广和应用有望提升渔业领域的保种技术水平,推动我国水产动物种质资源保存及管理工作。
综上所述,本软件包界面简约友好,功能较为全面,预期可以应用在水产动物种质资源保存管理、土著鱼类野生种群修复效果评价等很多领域。本软件提供的基因组近交分析工具可以协助建立基于水产动物遗传资源濒危程度评价模型,计算水产动物同一群体内或不同群体间个体亲缘关系、近交系数、保种所需有效群体规模等,并在此基础上,设计小群体保种的合理交配策略。
