Carbon-halogen bond activation by a structurally constrained phosphorus(III) platform
2021-10-14PenglongWngQinZhuYiWngGuixingZengJunZhuCongqingZhu
Penglong Wng,Qin Zhu,Yi Wng,Guixing Zeng*,Jun Zhu*,Congqing Zhu,*
a State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering,Nanjing University, Nanjing 210093, China
b Kuang Yaming Honors School, Institute for Brain Sciences, Nanjing University, Nanjing 210093, China
c State Key Laboratory of Physical Chemistry of Solid Surfaces and Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), and Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
1 These authors contributed equally to this work.
ABSTRACT The σ-bond activation by main group element has received enormous attention from theoretical and experimental chemists.Here, the reaction of C-X (X=Cl, Br, I) bonds in benzyl and allyl halides with a pincer-type phosphorus(III) species was reported.A series of structurally robust phosphorus(V)compounds were formed via the formal oxidative addition reactions of C-X bonds to the phosphorus(III)center.Density functional theory calculations show that the nucleophilic addition process is more favorable than the direct oxidative addition mechanism.Isomerization of bent structures of phosphorus(III) compound to poorly nucleophilic compounds to undergo further C-X bond activation can be rationalized by frontier molecule orbital analysis.This study not only provides a deep understanding of the reactivity of phosphorus(III) species but also demonstrates a potential of main group elements for the small-molecule activation.
Keywords:Pincer ligand Phosphorus C--X bond activation Main group element
Neutral tri-coordinate phosphorus (σ3-P) compounds are phosphorus compounds with a pyramidal structure, and have received significant attention due to their wide applications in synthetic chemistry [1-4].Owing to the facile regulation of the steric and electronic properties,the lone pair of electrons and the π-accepting properties of the σ3-P compounds have played important roles as nucleophilic catalysts [5,6].In recent years,distorted geometry-constraining tridentate pincer-type phosphorus compounds have received great attention because of their ability to cleave covalent chemical bonds [7].Research has demonstrated that the nontrigonal geometries of pincer-type phosphorus compounds(I-IV in Fig.1)are capable of O--H,N--H,B-H or B-O bond activation[8-14].Significantly,rare examples of the catalytic hydrogenation of azobenzene and the hydroboration of imine have been realized by these novel tri-coordinate phosphorus catalysts [10a,e].Density functional theory (DFT)calculations have shown that the phosphorus atom and the ligand both have a synergistic effect in this process [15].Despite such remarkable progress in this area, the oxidative addition of a C-X bond to the phosphorus atom remains underdeveloped.Experiments show that C-Br bond activation in benzyl bromide can not occur in the presence of compound IV because of the poorly nucleophilic character [10c].However, we herein report that a series of C(sp3)-X(X=Cl,Br and I)bonds in benzyl and allyl halides are activated by III via formal oxidative addition to the phosphorus center,leading to the formation of structurally robust phosphorus(V) adducts that permit complete characterization.

Fig.1.Representative examples of pincer-type phosphorus compounds.
As shown in Fig.2, the reaction of benzyl iodine with phosphorus(III) compound 1 in toluene solution proceeds slowly at room temperature,but after heating the mixture at 110。C for 2 days,the31P NMR spectrum indicates quantitative conversion of 1 to a new product,2a.The31P NMR spectrum of 2a shows a triplet resonance at δ -58.6 (2JPH= 14.6 Hz), which collapses to a singlet upon proton decoupling.Furthermore,a doublet was observed at δ 4.27 in the1H NMR spectrum,which was attributed to the protons in the CH2of the benzyl.The structure of 2a was further confirmed by single-crystal X-ray diffraction.Under the similar conditions,other substituted benzyl iodine species,such as 4-methoxybenzyl iodide and ethyl alpha-iodophenylacetate, have been observed to react with 1 to give the corresponding penta-coordinate phosphoranes 2b and 2c, respectively, for which phosphorus resonances were observed at-57.4 and-62.4 ppm as a triplet and a doublet in their31P NMR spectra, respectively.
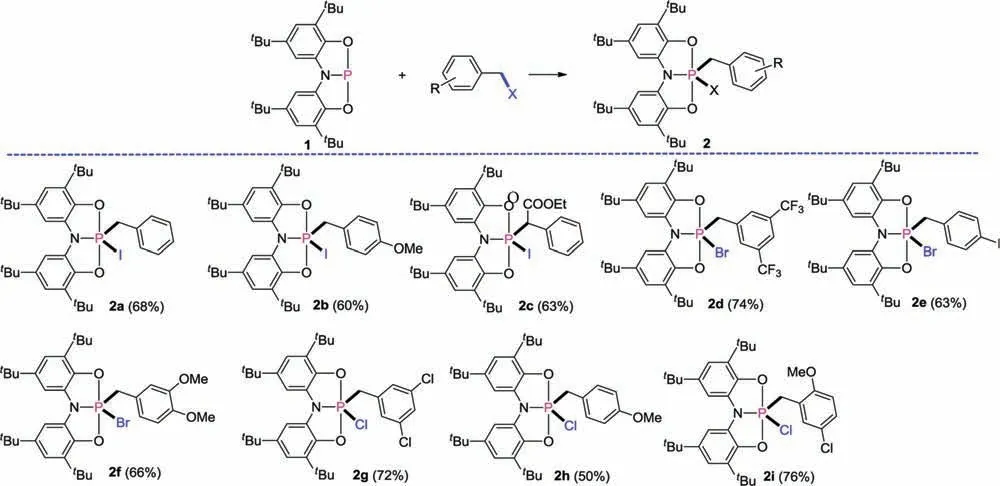
Fig.2.Reactions of compound 1 with benzyl halides.The isolated yields are given in parentheses.
The activation of benzyl iodines by 1 encouraged us to investigate its reactivity towards other benzyl halide substrates.The benzyl bromide and benzyl chloride substrates were also found to undergo formal oxidative addition with 1 to give 2d-2i(Fig.2).The presence of electron-withdrawing (2d) or electrondonating(2f and 2h)groups on benzyl did not inhibit the reactivity.Interestingly, only the C-X bonds of the benzyl were activated when aryl C-X bonds were contained in the same substrate(2e,2g and 2i).The resonances of the31P NMR spectra of the products were observed within a narrow range (-25.8 ~-21.7 ppm for benzyl bromide and-9.7 ~-6.8 ppm for benzyl chloride),and all of them collapse to a singlet upon proton decoupling, indicating that all adducts have a homologous structure.
Themolecularstructuresof2a,2e and 2g were furtherconfirmed by single-crystal X-ray diffraction.As shown in Fig.3, the ONO ligand exhibits a planar conformation,and the phosphorus center adopts a trigonal bipyramidal geometry in all these compounds,with O1 and O2 occupying the axial vertices(τ=0.69,0.74,and 0.67 for 2a,2e and 2g,respectively)[16].The bond angles of O1-P1-O2 in these series (171.1(3)。-175.66(1)。) were significantly larger than thatobservedincompound1(109.55(5)。)[11a].Thebondlengthsof P1-N1(1.696(3)-1.709(7)Å)in all species were clearly shorter than thatobservedincompound1(1.757(1)Å).However,bothP1-O1and P1-O2(1.684(6)-1.705(1)Å)aresignificant longerthanthose found in compound 1(1.659(1)and 1.652(1)Å,respectively).The lengths of the P1-C1 bond in these compounds arealmost identical,ranging from 1.823(3)Å to 1.830(3)Å.The bond lengths of P1-X1 for 2a,2e and 2g were 2.474(7),2.235(2)and 2.059(1)Å,respectively,which are consistent with the decreasing atomic radius from I to Br to Cl.The solid-state structures clearly show the formal oxidation additionproducts from compound 1 with benzyl halides.Therefore,the demonstration of C-X bond activation by a pincer-type phosphorus center was achieved by the reaction of benzyl halides with compound 1.
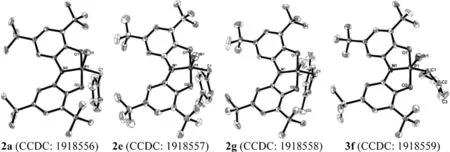
Fig.3.Molecular structures of 2a, 2e, 2g and 3f (thermal ellipsoids shown at 50% probability).Hydrogen atoms and solvents were omitted for clarity.
However, despite continued efforts, we could not activate the C-F bond by compound 1,which is probably due to the fact that the C-F bond has the highest bond dissociation energy in the series of C-X bonds [17].This result was consistent with the DFT calculations, where the t-butyl groups on the ONO ligand were replaced with methyl groups to reduce computational costs(Fig.4).We found that reaction barriers are too high to activate C-F bonds for both the direct oxidative addition and nucleophilic addition mechanisms.For the activation of other C-X bonds, our studies revealed that the nucleophilic addition pathways are preferred via Lewis acidic σ4-P+intermediate, leading to the formation of penta-coordinate (σ5-P) final product.Specifically,the reaction barriers for the activation of C-I,C-Br and C-Cl are 17.8,20.8 and 25.5 kcal/mol, respectively, which are much lower than those via direct oxidative addition pathways (35.7, 36.4 and 41.6 kcal/mol, respectively).The lowest reaction barrier for the activation of C-I bond was consistent with the experimental conditions with lower temperature.Moreover, transformation of Int1 to the final product is an intermolecular process (Fig.S1 in Supporting information).
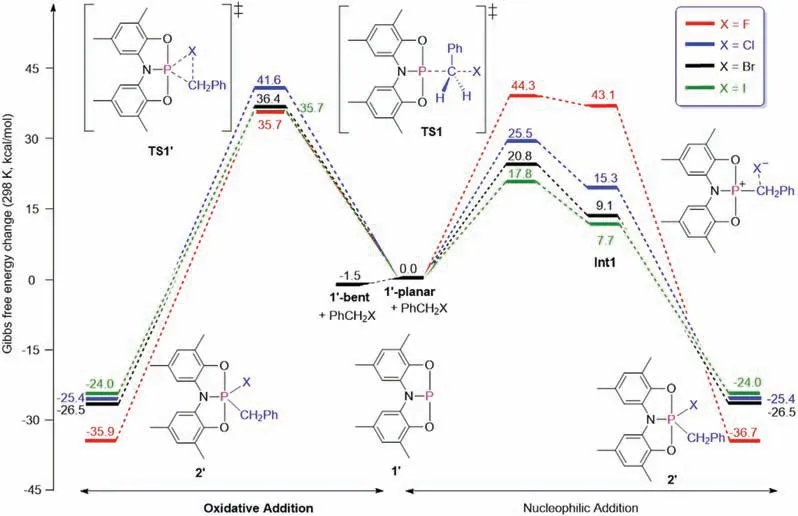
Fig.4.Gibbs energy profiles for the activation of C--X bonds by compound 1.
Surprisingly, we found this reaction was initiated by the strained geometry 1′-bent transforms to the C2vsymmetric planar geometry 1′-planar with endothermicity of 1.5 kcal/mol.However,according to natural bond orbital(NBO)analysis[18],the lone pair in the phosphorus-based compound 1′-bent and 1′-planar are in a hybridization of sp0.44and sp0.19, respectively, demonstrating the poor nucleophilicity of 1′-planar.In order to probe the origin of reaction mode by 1′-planar, we examined the frontier orbitals of 1′-bent and 1′-planar.As shown in Fig.5, the p orbital of phosphorus in 1′-bent mainly resides in HOMO-4 (-8.08 eV),whereas the p orbital of phosphorus in 1′-planar mainly resides in HOMO-1 (-6.39 eV).Therefore, the HOMO orbital in 1′-planar could be one of the major factors contributing to the C-X bond activation.
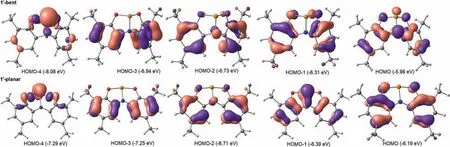
Fig.5.Occupied key molecular orbitals for 1′-bent and 1′-planar.
Further DFT calculations show that the activation of C-X bonds in allyl halides by compound 1 is also possible (Fig.S3 in Supporting information).The reaction barriers for the activation of allyl halides are slightly higher than those of benzyl halides,which was further confirmed by the energy decomposition analysis(EDA)of the transition states [19].In general, the reaction barrier contains the deformations of the reactants (ΔEDeform(1′) and ΔEDeform(C-X))and binding of the deformed reactants(ΔEb);thus ΔE(barrier) = ΔEDeform(1′) + ΔEDeform(C-X) + ΔEb.As shown in Fig.6,the binding energy(ΔEb)provides the major contribution to the difference of those reaction barriers.
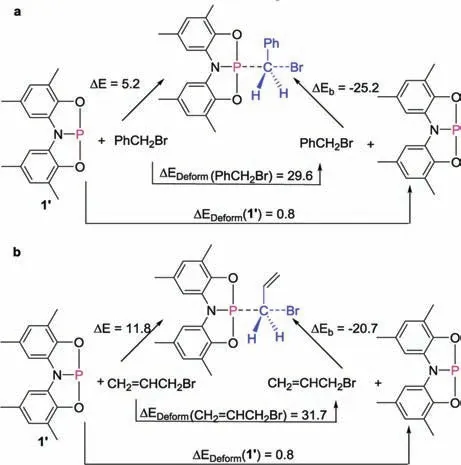
Fig.6.EDA on the transition states of C-Br activation by 1′.The energies are given in kcal/mol.
Therefore, treatment of compound 1 with allyl iodide leads to the formation of the C-I bond cleavage product 3a (Fig.7).NMR showed that compound 1 was fully consumptive, leading to a characteristic penta-coordinate phosphorus resonance observed at δ -58.1 in the31P NMR spectrum, which is consistent with the values observed for 2a-2c(ranging from-62.4 ppm to-57.4 ppm).Similar to compound 2, this signal showed a proton coupled multiplet that collapsed to a singlet after proton decoupling.The signal for the CH2protons of the allyl group in 3a was observed at 3.59 ppm as a doublet due to the coupling of the phosphorus.The C-I bonds in other allyl iodides with different substitutes(3b and 3c in Fig.7) were also reacted with compound 1.
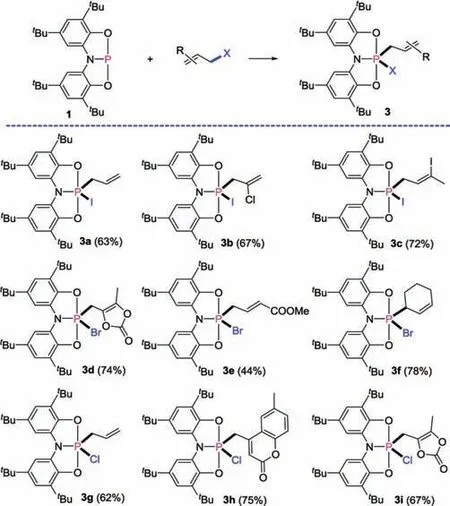
Fig.7.Reactions of compound 1 with allyl halides.The isolated yields are given in parentheses.
Under similar conditions,allyl bromide and allyl chloride were also found to undergo formal C-X bond oxidative addition with 1.As shown in Fig.7, the substituted allyl bromide, such as 4-bromomethyl-5-methyl-1,3-dioxol-2-one, methyl 4-bromocrotonate, and 3-bromocyclohexene, could react with compound 1 to form penta-coordinate phosphoranes 3d-3f in high yields.The31P NMR resonances in each species were observed in the narrow range of -26.5 to -13.6 ppm, consistent with an analogous structure of penta-coordinate adducts 2d-2f(-25.8 ~-21.7 ppm).In addition, the C-Cl bonds in allyl chloride with different substitutes or heterocycles (3g-3i) were also reacted by the phosphorus center in compound 1.The proton-coupled31P NMR resonance(-12.1 ~-7.6 ppm)was also observed for these adducts of allyl chloride with compound 1.
One of the solid-state structure of these products, 3f was further confirmed by single-crystal X-ray diffraction studies(Fig.3).The general features on the phosphorus center were similar to those observed in compounds 2a, 2e and 2g.The bond lengths of P1-C1 and P1-Br1 were 1.840(3) Å and 2.257(8) Å,respectively, which are slightly longer than those found in 2e.These results confirm the formal oxidative addition of C-Br bonds to the phosphorus(III) center in compound 1.
We concluded that the geometrically constrained phosphorus(III)compound 1 can react with the C-X bonds(X=Cl,Br and I)in benzyl halides and allyl halides,leading to the formation of pentacoordinate adducts.DFT calculations suggest that the nucleophilic addition pathway is more feasible than the direct oxidative addition mechanism.This study further demonstrates that pincertype phosphorus(III) can maintain the nucleophilic character,highlighting the wide application of geometrically constrained phosphorus(III) as an effective reagent.
Declaration of competing interest
The authors declare no competing financial interests.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (Nos.21772088 and 21573179), the Natural Science Foundation of Jiangsu Province (No.BK20170635), the Young Elite Scientist Sponsorship Program of China Association of Science and Technology, the program of Jiangsu Specially-Appointed Professor and Shuangchuang Talent Plan of Jiangsu Province.
Appendix A.Supplementary data
Supplementary material related to this article can be found,in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.005.
杂志排行

Chinese Chemical Letters的其它文章
- Recent development of pillar[n]arene-based amphiphiles
- Recent advances in synthesis of organosilicons via radical strategies
- High thermal conductivity of graphene and structure defects:Prospects for thermal applications in graphene sheets
- Binder-free electrodes for advanced potassium-ion batteries:A review
- Sulfone-based high-voltage electrolytes for high energy density rechargeable lithium batteries: Progress and perspective
- Recent advances in fluorescence imaging of alkaline phosphatase