Mechanism of the Ir/Pd catalyzed photocarboxylation of aryl halides
2021-10-14YingLvBingWngHizhuYu
Ying Lv,Bing Wng*,Hizhu Yu,*
a Department of Chemistry and Centre for Atomic Engineering of Advanced Materials, Anhui University, Hefei 230601, China
b Department of Chemistry, University of Science and Technology of China, Hefei 230026, China
ABSTRACT The recent Ir/Pd co-catalyzed photo carboxylation of aromatic halides with CO2 has shown high efficiency and excellent functional group tolerance for preparing aromatic carboxylic acids and esters.With the aid of density functional theory (DFT) calculations, the carboxylation starts with two parallel steps, i.e.,oxidative addition of aromatic halides on Pd0 and reductive quenching of the photocatalyst Ir(ppy)2(dtbpy)+with amine.Thereafter,a successive oxidation of PdII with the amine radical(generated by the reaction of cationic radical amine and Cs2CO3)and IrII species occurs to generate Pd0,from which the carboxylation occurs easily via a coordination, Pd-C insertion step.The release of the carboxylate product then regenerates the catalyst.
Keywords:Carboxylation Pd-catalysis DFT Charge state Mechanism
As an abundant, readily-available, green and renewable C1 block, CO2has recently attracted increasing research interest[1-12].The carboxylation of organometallic reagents (such as Grignard reagent and organic lithium reagent) with CO2has been reported for decades [13-16].Despite the great success,these reactions always suffer from the drawbacks such as poor selectivity and harsh reaction conditions(sensitive to water and oxygen).With the development of transition metal catalysts(such as Pd, Ni and Co complexes), highly selective carboxylation with CO2have been reported [17-19], using the relatively mild metal/metalloid reagents, such as organotin [20-23],organozinc [24,25] and organoboron [26-32], as substrates.
A breakthrough on direct carboxylation of aromatic halides with CO2under mild conditions (40。C) was recently reported by Martin and co-workers, using Pd/tBuXPhos (tBuXPhos=2-di-tertbutyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine) as the catalyst(Scheme 1a)[33].Thereafter,a variety of transition metal(such as Ni,Cu)catalysis has been developed,and greatly expands the functional group tolerance of the carboxylation[34-36].Nevertheless, the used metal reducing agents, such as Et2Zn and Zn/Mn powder,result in the necessity for post treatment of the metal wastes.
To improve the robustness of the carboxylation reactions,chemists recently developed a series of novel photocarboxylation reactions.For example,Iwasawa and co-workers recently reported the dehalogenation-carboxylation of aromatic halides with the combination of the photocatalyst Ir(ppy)2(dtbpy)(PF6) (ppy:2-phenylpyridine; dtbpy: 4,4′-di-tert-butyl-2,2′-dipyridyl) and the transition metal catalyst Pd(OAc)2(Scheme 1b) [37], in the presence of the additive N,N-diisopropyl ethylamine and PhXPhos/tBuXphos (PhXPhos: 2-diphenyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphine).The carboxylation has shown excellent functional group tolerance, and gave the corresponding methyl ester derivatives (after the methyl esterification) in good yields.
The preliminary mechanistic probes by Iwasawa et al.suggests a catalytic cycle involving the reduction (Pd(OAc)2→Pd0), oxidative addition (of aromatic halide), carboxylation, dehalogenation and catalyst-regeneration steps [37].Albeit the reasonability of the proposed mechanism, some details remain to be clarified.For example,the direct reaction of PdIIwith CO2,or a prior reduction of PdIIto PdIor Pd0over the carboxylation are all possible(Scheme 2).In other words, the accurate valent state and structure of the precursor before the carboxylation step are still unknown.Meanwhile, the quenching mode of the photocatalyst (oxidative/reductive quenching) and how the photocatalyst cooperates with the transition metal catalyst are all unsettled problems.In this context,density functional theory(DFT)calculations were used to investigate the mechanism of the Ir/Pd co-catalyzed photocarboxylation of aromatic halides with CO2.
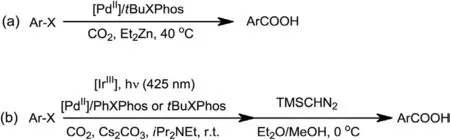
Scheme 1.Pd catalyzed dehalogenation-carboxylation of aromatic halides with CO2 reported by Martin et al.(a);and Ir/Pd co-catalyzed carboxylation of aromatic halides reported by Iwasawa et al.(b).
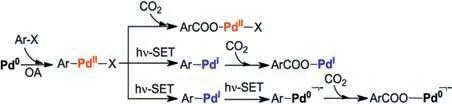
Scheme 2.The possible carboxylation pathways on Pd0/I/II species.
In this study,all calculations were performed with the Gaussian 16 software [38].The B3LYP functional [39,40] is used in the geometry optimization for all species.The effective core potential and the associated basis set of SDD[41]is used for Ir and Pd atoms,and the 6-31G(d,p)basis set is used for other atoms(including C,H,O, N, P, Br).Frequency analysis was performed at the same level with geometry optimization to ensure the number of imaginary frequencies (1 for transition state and 0 for other species), and to obtain the thermal correction to Gibbs free energy.Single point energy calculations were carried out at a higher level of theory:B3LYP/def2-TZVP [42].In all these calculations, the dispersion correction reported by Grimme and co-workers (i.e., Grimme-D3BJ) was used [43], and N,N-dimethylacetamide was used as solvent(in accordance with Iwasawa’s experiments[37],and with SMD model [44]).Natural bond orbital (NBO) [45] analysis was carried out at the same level with geometry optimization.The optimized structures were plotted by Cylview software [46].
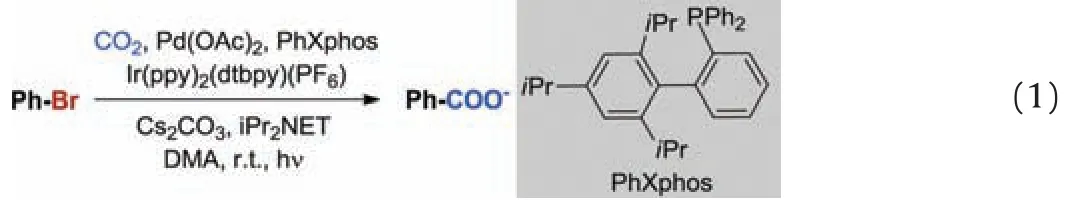
Bromobenzene was chosen as the modeling reactant due to its structural simplicity and the high conversion efficiency.To this end,the dehalogenation and carboxylation of bromobenzene with CO2in the presence of Ir/Pd co-catalysts and PhXphos was used as model reaction (Eq.1).
Our calculations start with the PhBr and PhXphos coordinated Pd0intermediate I1 (Scheme 3).For clarity, I1 was set as the energetic reference point.From I1,oxidation addition occurs easily via the typical three-membered ring transition state of TS1(barrier:5.4 kcal/mol), and the relative energy of the formed I2 is indicate that the oxidative addition is favored from both thermodynamic and kinetic aspect.Of note, I2 was also verified by the experimental observations by Iwasawa and co-workers[37].
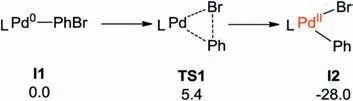
Scheme 3.Energy changes for the oxidative addition step (in kcal/mol, L=PhXphos).
From I2, we first examined the possibility for its direct carboxylation with CO2.As shown in Fig.1, the approaching of CO2to I2 generates the intermediates I3, from which the direct carboxylation occurs via the transition state TS2 (Figs.1 and 2).
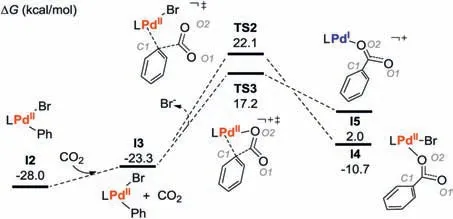
Fig.1.Pd catalyzed reduction carboxylation (kcal/mol).
The energy barrier for the transformation of I3→TS2 is as high as 45.4 kcal/mol, presumably due to the high steric hindrance around the metal center(Fig.2),the low nucleophilicity of aryl C1 atom (Fig.1, with NPA charge of -0.081) and the Zwitterionic character of the transition state (NPA charge of O1/O2 atom is-0.648/-0.678) in TS2.Alternatively, a pre-dissociation of bromide could provide a vacant coordination site on the palladium center,making it possible to accommodate the dangling O2 atoms to reduce steric hindrance and avoid the formation of the Zwitterionic intermediates (TS2 vs.TS3 in Fig.2).To this end,the energy barrier of the carboxylation step was decreased to 40.5 kcal/mol.Nevertheless, the energy demand of over 40 kcal/mol remain too high to occur under the experimental conditions.Therefore, the results herein indicate that the main difficulty in the PdIIparticipated carboxylation lies in the low electron density of the aromatic group.In this context, we examined the possibility for a pre-reduction of the carboxylate precursor of I2.
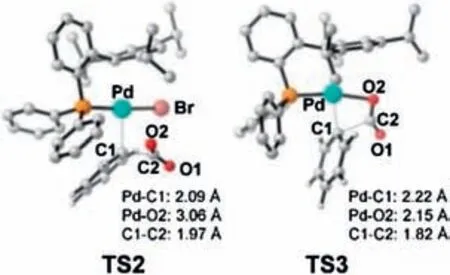
Fig.2.The optimized geometries of TS2 and TS3.H atoms are omitted.
I2 could be reduced by either the excited state of photocatalyst(i.e.,IrIII*),or the reduced photocatalyst(IrII,vide supra)generated from the catalytic cycle of the photocatalyst.Therefore,the relative facility of the reductive and oxidative quenching of the photocatalyst was examined [47-50].In oxidative quenching, IrIII* is oxidized by I2 to generate IrIVand I6 (Scheme 4), while in the reductive quenching, IrIII* is reduced by Amine1 to generate Amine2 and IrII.According to the calculation results,the reductive quenching is exergonic by 15.4 kcal/mol,and is more feasible than the oxidative quenching(endergonic by 2.6 kcal/mol).In addition,considering the existence of Cs2CO3, the removal of H+from Amine2(formed during the reductive quenching of photocatalyst)could occur easily to generate the radical [iPrNEt(CMe2)]·(Amine3).In this context, Amine3 could possibly act as the electron donor to reduce I2.This process is exergonic by 1.7 kcal/mol (Scheme 4), and generates the cationic [iPrNEt(CMe2)]+(Amine4)and the I6.By contrast,the reduction of I2 with IrII(IrII+I2→IrIII+ I6) or IrIII*mentioned above is endergonic by 6.4 and 2.6 kcal/mol (Scheme 4), and is thus less favorable.
From I6,the dissociation of Br-occurs easily,generating a more stable,neutral intermediate I7.According to the calculation results,the released Br-is unlikely to bind with the cationic Amine4 to form contact ion pair or neutral complex(Scheme S1 in Supporting information).From I7, either a direct carboxylation(I7→I8→TS4→I9,Fig.3)or a reduction[37]-carboxylation pathway(I7→I11→I12→TS5→I10) might occur (the reduction-dehalogenation pathway on I7 was excluded due to the high energy demand for formation of the dianionic I13, Fig.3).
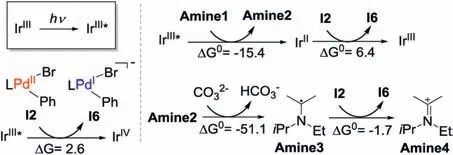
Scheme 4.The reduction of PdII intermediate I2 (kcal/mol).
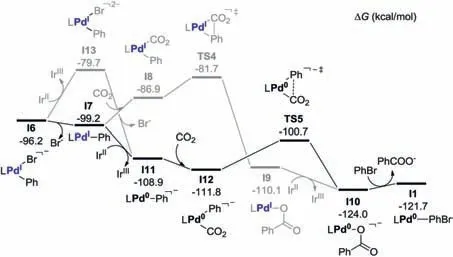
Fig.3.Energy profiles for the possible carboxylation steps on I6 (in kcal/mol).
In the first case, the approaching of CO2first generates the intermediate I8,and this step is endergonic by 12.3 kcal/mol.In I8,both the short Pd-C2 distance(2.19 Å in Fig.4)and the Mayer bond order (0.44) indicate the pre-activation of CO2.In the formed intermediate I9,the carboxylate product mainly coordinates with the palladium center via the terminal O2 atom(Fig.4).The energy barrier for the direct carboxylation on I8 is 4.8 kcal/mol, and the relative Gibbs free energy of the formed I9 is lower than I8 by 23.2 kcal/mol.The remarkably lower energy barrier for the carboxylation step of I8 than that of I3 (5.2 vs.45.4 kcal/mol)further evidences the importance of the enhanced nucleophilicity on the carboxylation step(NPA charge of the aryl C1 atom in I8 and I3 is-0.384 and-0.085).After that,the one electron reduction of I9 could occur to generate a more stable, Pd0intermediate I10.Finally, the formed Pd0intermediate I10 could undergo a ligand exchange to release the carboxylate product and regenerate I1.
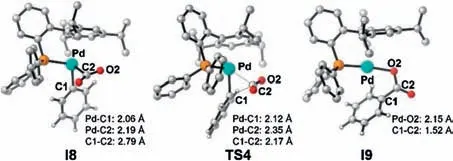
Fig.4.The optimized geometries of I8, TS4 and I9.H atoms are omitted.
In the second case,the reduction of I7 with IrIIis exergonic by 9.7 kcal/mol.Due to the high electron density on the Pd0center of I11 (NPA charge on Pd is -0.540), the coordination of CO2occurs easily via an η1-C coordination mode (exergonic by 2.9 kcal/mol),while the coordination of CO2by an end-on mode is unfavorable(Scheme S2 in Supporting information).The Pd-C1 distance in I12 is 2.09 Å (Fig.5), and its Mayer bond order of 0.71, indicating the formation of σ(Pd-C1) bond therein.Such intermediate is typical for the interaction of CO2with the electron rich metal complexes[51].Thereafter, the C--C bond formation was achieved via the carboxylation transition state TS5, with a low energy barrier of 11.1 kcal/mol.The Pd0intermediate I10 is then generated as the carboxylate product, and the subsequent transformation is the same as the aforementioned carboxylation-reduction pathway.
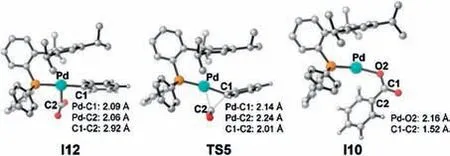
Fig.5.The optimized geometries of I12, TS5 and I10.H atoms are omitted.
Comparing the different pathways in Figs.1 and 3, the most feasible carboxylation pathway involves the cascade reduction of the PdIIintermediate to Pd0, associating with the release of bromide (I2→I6→I7→I11).Then the coordination of CO2, carboxylation, and ligand exchange occurs to release the carboxylate product (I11→I12→I10→I1).In combination with the calculation results in Schemes 3 and 4, the rate determining step for the dehalogenation-carboxylation mechanism is the elementary carboxylation step, and the overall activation free energy barrier is 11.1 kcal/mol.The low energy barrier is in good agreement with the high efficiency of Iwasawa’s reaction under room temperature[37].More importantly,the easiness of the elementary carboxylation step was found to correlate with the nucleophilicity of the Pd precursor.According to the distortion-interaction analysis on the three key carboxylation transition states, i.e., TS3 (PdII), TS4 (PdI)and TS5(Pd0),the interaction energies between CO2and the other part in the target transition state decreases with reduced valence state of the Pd center(the interaction energy of TS3,TS4,TS5 was-22.4, -36.4, -53.4 kcal/mol, respectively), demonstrating the stronger interaction between the reduced Pd complex and CO2.The results verified the aforementioned proposal that the Pd complex with electron rich palladium center and aromatic groups are more prone to undergo a nucleophilic attack to CO2.Nevertheless,due to the remarkably stronger interaction between CO2and Pd0complex in TS5 (than that in TS3 and TS4), CO2distorted significantly compared to the free state(Scheme S3 in Supporting information),and thus the distortion energy of TS5 is significantly higher than TS3 and TS4 (49.5 vs.40.6, 40.8 kcal/mol).To this end, the energy barrier of the transition state TS4 with balanced the interaction and distortion energy is the lowest, while the weak interaction between the PdIIcenter and CO2results in the highest energy barrier of TS3.
In this study,the mechanism for the dehalogenation-carboxylation of aryl halides recently reported by Iwasawa and co-workers were systematically studied by DFT calculations.The overall mechanism consists of oxidative addition of aryl halides,excitation and reductive quenching of the IrIIIphoto-catalyst, successive reduction of PdIIto PdIand Pd0, removal of bromide, and carboxylation steps.The carboxylation with CO2is sensitive to the valence state of the palladium center,and thus the presence of reductant(i.e.,oxidized state of the photocatalyst IrIIand the amine radical) is pivotal to the facility of the carboxylation step.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
We appreciate the financial support from the National Natural Science Foundation of China(Nos.21672001,51961135104)and the technical support of high-performance computing platform of Anhui University.
Appendix A.Supplementary data
Supplementary material related to this article can be found, in the online version,at doi:https://doi.org/10.1016/j.cclet.2020.09.045.
杂志排行

Chinese Chemical Letters的其它文章
- Recent development of pillar[n]arene-based amphiphiles
- Recent advances in synthesis of organosilicons via radical strategies
- High thermal conductivity of graphene and structure defects:Prospects for thermal applications in graphene sheets
- Binder-free electrodes for advanced potassium-ion batteries:A review
- Sulfone-based high-voltage electrolytes for high energy density rechargeable lithium batteries: Progress and perspective
- Recent advances in fluorescence imaging of alkaline phosphatase