二硫化钼自组装微球的制备及其吸附性能
2021-10-14唐庭志孙元平邵建华
唐庭志,孙元平,钱 宏,邵建华
(烟台大学光电信息科学技术学院,山东 烟台 264005)
二硫化钼(MoS2)是一种新兴半导体材料,稳定的2H-MoS2为六方层状结构晶体,每个钼原子连接6个硫原子,形成了S-Mo-S状的三明治结构,层与层之间通过微弱的范德华力结合,使得MoS2在润滑领域得到广泛的应用[1]。同时,单层MoS2是禁带宽度为1.9 eV的直接带隙半导体,这也让MoS2在光电器件领域有广泛的应用前景[2]。依据生长条件不同,MoS2可呈现出多种不同形貌,如镂空网状、类花状、块状[2]等。目前单层MoS2的生长与特性研究已经很多,如光学、力学等[3-6],而水热制备得到MoS2中空微球的生长及吸附研究则相对较少。从已有的报道中可知,MoS2纳米片在软模版上可以通过自组装而形成球状结构,可增加其比表面积并形成独特的介孔结构,使得MoS2在污水处理、有机吸附领域有广泛的应用前景[7-8]。
本文通过微波辅助水热法合成了MoS2中空微球结构,并研究了微球生长机理及形貌特征。亚甲基蓝(MB)溶液的吸附性能测试表明,相比于片状结构, MoS2微球对于MB溶液的10 min吸附量得到了很好的提升,使得MoS2有望成为新一代高性能吸附剂。
1 实验
MoS2的制备分别以钼酸钠(Na2MoO4)、硫代乙酰胺(CH3CSNH2)作为钼源和硫源,十六烷胺甲基四胺(CTAB)作为表面活性剂以及生长模板,去离子水(H2O)为溶剂。实验所用的药品均为分析纯级,未进一步提纯。
将4 mmol Na2MoO4和12 mmol CH3CSNH2(Mo∶S为1∶3)放入100 ml烧杯中并加入35 ml去离子水,电磁搅拌10 min待溶质完全溶解后,再加入0.05 g CTAB并继续搅拌10 min。将均匀混合溶液倒入内胆为聚四氟乙烯的水热合成釜中并升温至210 ℃,反应过程中最大压强为5 MPa,反应1.5 h后自然冷却至室温。将反应得到的黑色样品利用去离子水和无水乙醇超声清洗3次后,在70 ℃ 下烘干6 h,得到黑色粉末样品,标记为CM1∶3-1.5;在同样的生长条件下,反应时间为2 h、2.5 h得到的样品标记为CM1∶3-2、CM1∶3-2.5;钼硫比为1∶2、1∶5而反应时间为2.5 h制备得到的样品标记为CM1∶2-2.5、CM1∶5-2.5;无CTAB添加制备得到的样品标记为CM0。
利用日本理学株式会社(Rigaku)的SmartLab III型X射线衍射仪进行XRD表征;日本株式会社(JEOL)型号为JSM-5610V的扫描电镜进行SEM表征;英国雷尼绍(Renishaw)型号为Via-Reflex激光共聚焦拉曼光谱仪,激发光源为波长532 nm的掺钕钇铝石榴石激光器进行Raman光谱表征。贝士德仪器科技(北京)有限公司型号为BSD-PM1的高性能比表面积及微孔分析仪测试BET比表面积。上海添时科学仪器的型号为EU-2800D的紫外可见光分光计测试吸光度。
2 结果与分析
2.1 XRD
图1给出了CM1∶3-1.5、CM1∶3-2、CM1∶3-2.5样品的XRD图,通过卡片检索可以看出,样品的衍射峰与JCPDS.37-1492相符。主要衍射峰13.77°、33.1°、39.07°、58.95°分别对应六方晶相MoS2的(002)、(100)、(104)、(110)晶面,且没有出现其他衍射峰,说明实验得到的黑色粉末为较高纯度的2H-MoS2。
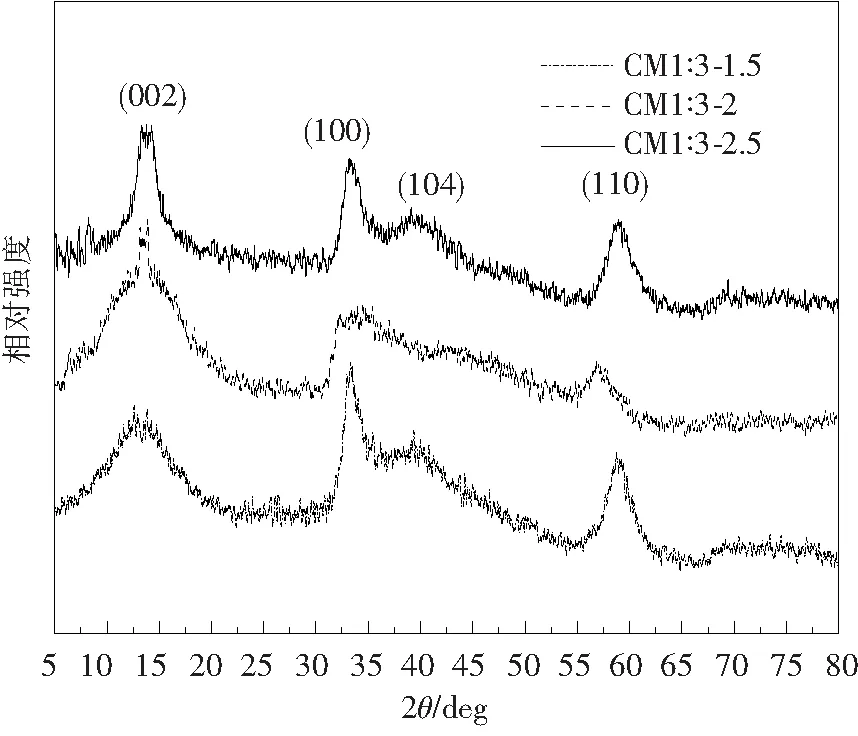
图1 不同反应时间下生长的MoS2样品的XRD谱
(002)晶面为平行于MoS2片层方向的晶面,是由MoS2层与层堆叠结构产生的衍射。随着反应时间的增长,(002)衍射峰的半高宽明显变窄,说明反应时间增长有利于MoS2层状堆叠结构的生长,其中MoS2单个片层结构的结晶度较高[9]。而(100)、(110)衍射峰则主要是由于MoS2层内结构(S-Mo-S三明治结构)产生的衍射,这2个峰的出现也证明了得到的MoS2纯度较高。CM1∶3-2样品的(100)衍射峰较弱可能是由于CTAB插层导致MoS2层内结构被破坏。
2.2 样品的SEM分析
为了对生成样品的表面形貌进行表征,进行了SEM测试,不同钼硫比生长条件下样品的SEM图如图2所示。从图2(a)、(b)可以看出,在CTAB辅助下制备得到的MoS2呈现出分散性良好且较为规则的球状结构。
在图2中取30个微球测量其直径并取平均值,得到CM1∶3-2.5微球直径约为1.5 ± 0.5 μm,而CM1∶2-2.5微球直径约为0.9±0.3 μm,CM1∶5-2.5微球直径约为500±100 nm,说明当钼硫比小于或者大于1∶3时,微球尺寸均有不同程度的减小。当钼硫比为1∶5时,生成的MoS2微球出现团聚现象,主要由于纳米颗粒的表面效应,随着MoS2微球尺寸减小,表面原子比例迅速增加,使得表面原子处于高活化状态极易与其他原子结合,导致团聚现象。图2(c)为CM1∶5-2.5样品的SEM图,表明该微球结构由MoS2纳米片聚集而成,在片与片之间存在大量的微孔。图2(e)为CM1∶3-2.5样品的单个破损球SEM图,图2(f)为CM1∶3-2.5样品的完整微球结构的SEM图。从图中可以看出,这种微球结构的内部中空,球壳厚度达到了120 ± 15 nm左右,而微球表面则由大量MoS2纳米片堆叠而成,这种纳米片的堆叠也让表面呈现了一个多孔隙的结构[10]。对于破损的微球,中空的结构使得微球内部也有着吸附作用,可以提高单位质量样品的吸附量以及吸附效率,让其在吸附领域有更大的潜能[11-12]。图2(d)给出了CM0样品的SEM图,可以看出生成的MoS2样品为大量MoS2纳米片堆叠而无定型,说明CTAB在MoS2成球过程中起到了重要作用。
2.3 Raman光谱
图3给出了CM1∶3-2.5、CM1∶2-2.5、CM1∶5-2.5样品的室温Raman光谱图。2H-MoS2的Raman峰有2个,分别是E2g1(383 cm-1)以及A1g(408 cm-1),其中E2g1对应着2H-MoS2中的钼原子与硫原子沿着平层面方向振动,而A1g则对应着2H-MoS2的钼硫键在垂直方向上的层间振动。E2g1和A1g2种振动模式的峰位一定程度上受MoS2片层厚度影响,随着片层厚度增加峰位差增加[13]。从图2中可以看出合成的样品在379 cm-1和402 cm-1左右均有拉曼峰,证明合成了2H-MoS2。当硫钼比为1∶3时,样品的2种振动模式峰位差最小为22 cm-1,小于文献[13]中介绍的25 cm-1峰位差。硫钼比1∶2或1∶5条件下生成的样品,峰位差过大,主要是由于生长过程中,过量的S或者Mo导致c轴方向片层晶体结构缺陷[13]。

图2 不同硫钼比条件下MoS2样品的SEM图
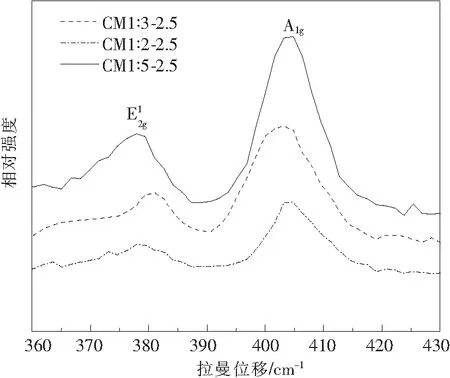
图3 不同钼硫比制得的MoS2样品的Raman光谱
2.4 样品BET分析
比表面积大小是影响吸附性能的重要因素。图4(a)、(b)分别是CM1∶3-2.5、CM0样品的氮气吸脱附等温线。两样品的氮气吸脱附等温线均为Ⅳ型等温线,具有明显的迟滞环,CM1∶3-2.5样品的BET比表面积为32.76 m2/g,CM0样品的BET比表面积为18.62 m2/g。图4(a)中随着相对压力的增加,吸附发生在多层的外表面,当相对压力为0.9左右时,吸附量快速增加,这是由多孔狭缝吸附引起的[14]。图4(c)是CM1∶3-2.5样品的孔径分布曲线,在2 nm和13 nm处有主峰,可归属于纳米片间狭缝以及纳米片聚集而产生的大孔。图4(d)为CM0样品的孔径分布图,孔径主要集中在10 nm左右。最终表明生长得到的MoS2微球样品具有较高的比表面积以及良好的介孔结构。
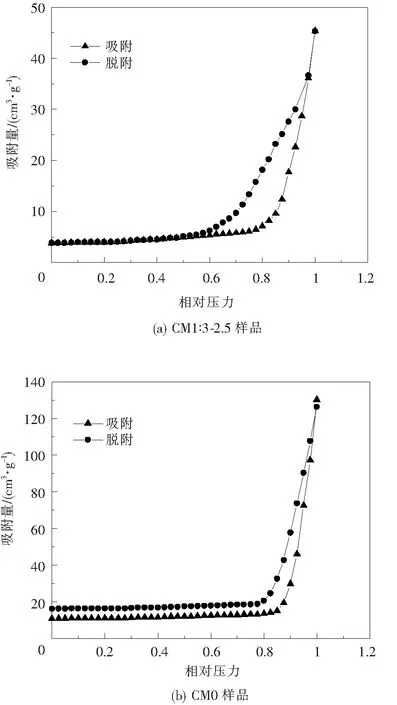
图4 MoS2样品的氮气吸脱附曲线及孔径分布
2.5 微球的形成机理
以简易微波水热方法制备MoS2,通过控制改变单一变量如:生长时间、钼硫比以及是否添加CTAB,制备得到多组样品。XRD图谱以及Raman图表明合成了高纯度的2H-MoS2,并得出钼硫比为1∶3,210 ℃条件下反应2.5 h,生长的2H-MoS2结晶度最好。SEM测试结果表明,CTAB在微球生长过程中起到了重要作用,其生长机理如下:高温环境下,钼酸钠分解生成MoO3反应的前驱体,同时硫代乙酰胺水解生成H2S作为反应的还原剂。CTAB由(C4H9)4N+组成,阳离子在水中形成许多的小泡,其中疏水基团聚在一起形成核心,而亲水基团形成外壳,表面阳离子通过静电或者氢键作用吸引S2-,同时MoO42-受静电作用吸引在表面被还原生成MoS2,形成外部由大量纳米片自组装而成的空心微球(图5)。反应过程如下:
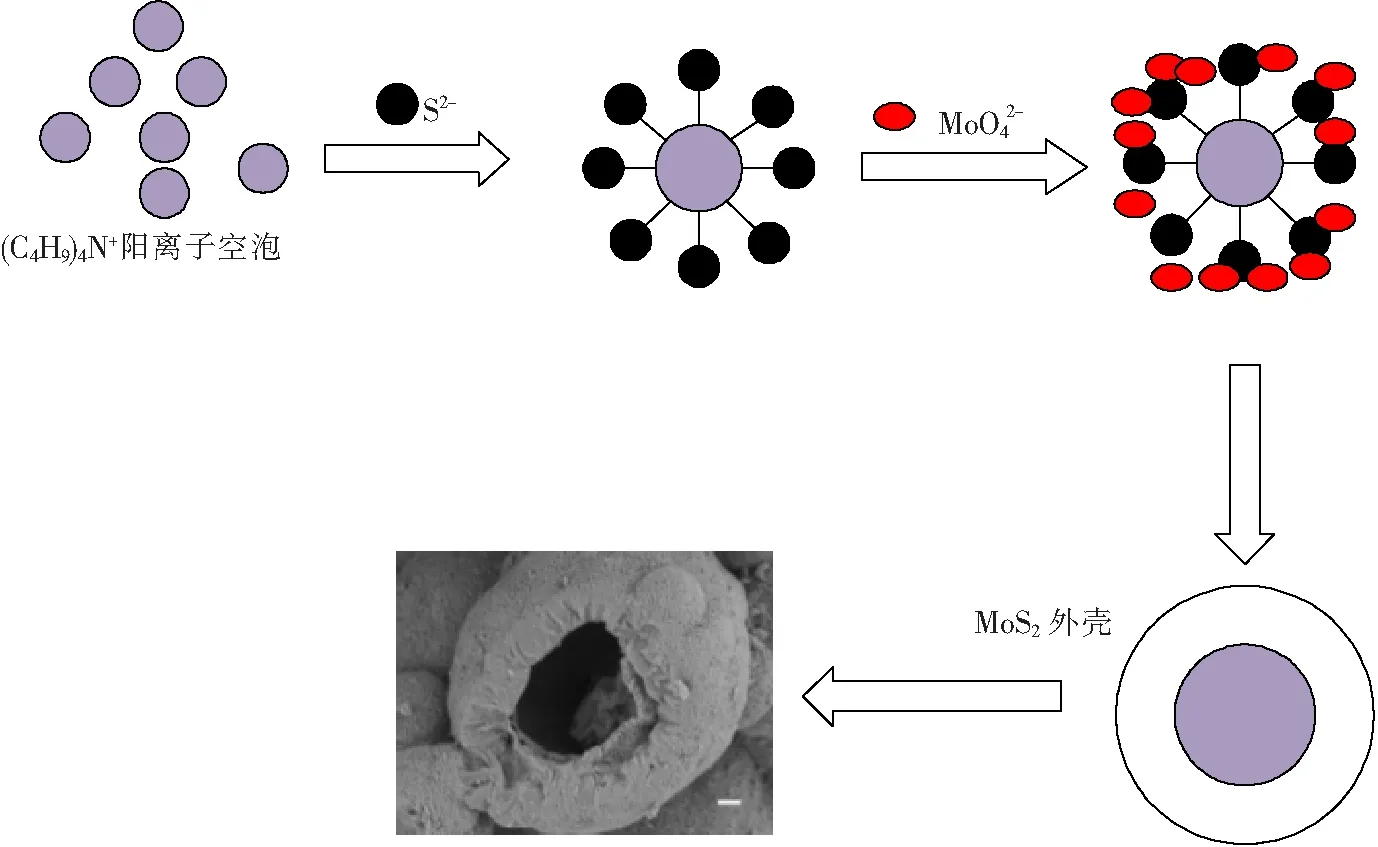
图5 MoS2样品生长机制
CH3CSNH2+2H2O→H2S+CH3COOH+NH3,
H2S→2H++S2-,
MoO42-+2S2-+8H+→MoS2+4H2O。
同时不同钼硫比情况下,微球尺寸不同主要是由于当钼硫比过大或者过小时,反应速率以及成核速率的加快是导致微球尺寸变小的主要原因之一[15]。比表面积大小对于吸附性能有着重要影响,微球形貌MoS2比表面积要大于团簇状MoS2纳米片,且均有介孔结构,较大的比表面积提供了更多的活性点,这使得MoS2微球的吸附性能更优于MoS2纳米片。
3 MoS2的吸附性能
3.1 吸附实验
为探究MoS2对有机染料的吸附性能,利用MB溶液进行了吸附实验,并且根据吸附动力学,对于介孔结构吸附剂,孔径过小并不利于吸附有机溶剂,所以利用CM1∶3-2.5和CM0样品进行实验。在27 ℃条件下配置不同质量浓度的MB溶液,分别为10、15、20、25、30 mg/L。分别取50 mL不同浓度的溶液置于烧杯中,加入0.03 g MoS2样品,将混合物置于多功能振荡仪充分振荡,震荡过程无光照,振荡1 min后取5 mL混合物,之后每隔2 min取5 mL混合物在3500 r/min条件下离心1 min,取上清液利用紫外-可见光分光计在MB溶液最大吸收波长664 nm处测量其吸光度[16],并根据MB标准曲线计算其浓度,图6给出10、8、5、1 mg/L的MB溶液的吸收光谱及标准曲线。
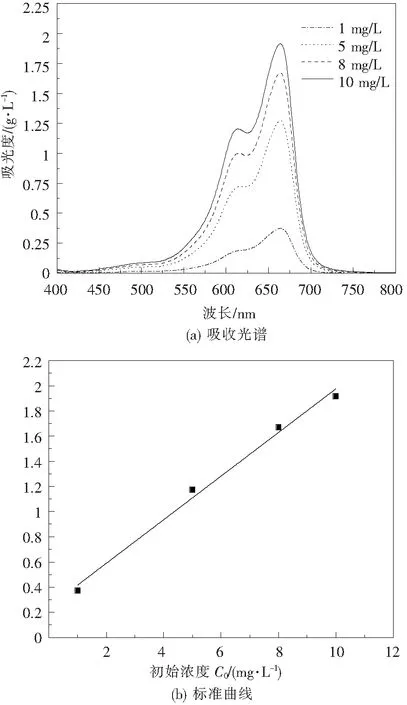
图6 MB溶液吸收光谱及标准曲线
利用以下公式计算其吸附量以及吸附速率[17]
q=(C0-Ct)×(V/M),
(1)
v=(C2-C1)×(V/T),
(2)
公式(1)可计算出在不同时刻MoS2对MB溶液的吸附量,其中,q表示在t时刻的吸附量,mg/g;C0表示MB溶液的初始质量浓度,mg/L;Ct表示在t时刻的MB溶液质量浓度,mg/L;V表示溶液体积,L;M表示吸附剂MoS2质量,g。公式(2)可计算出在一段时间内MoS2对MB溶液的吸附速率,其中v表示在一段时间内的吸附速率,mg/min;T表示2次测量的时间间隔,min。C2-C1表示相邻2次测量的MB溶液浓度差,mg/L。
3.2 亚甲基蓝溶液浓度对吸附的影响
在室温下,测试了0.03 g CM1∶3-2.5和CM0样品对不同质量浓度MB溶液的总吸附量,总吸附时间为10 min。取最终溶液用紫外-可见光分光计测量其浓度得如图7结果。
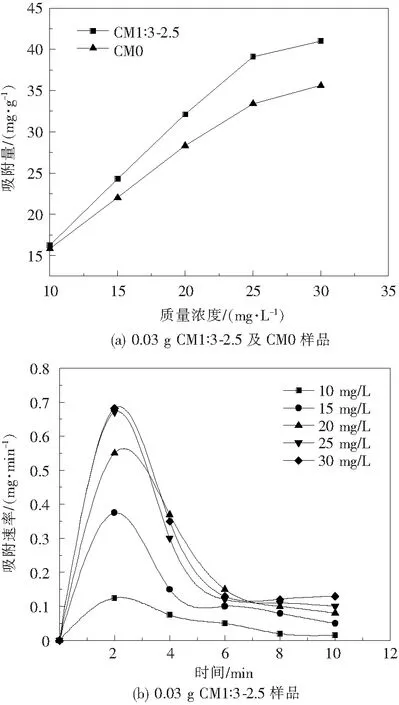
图7 MoS2样品对不同浓度溶液的吸附量及吸附速率
从图7(a)中可以看出,对于0.03 g CM1∶3-2.5和CM0样品,微球结构MoS2吸附量始终大于MoS2纳米片,说明微球结构确实提高了MoS2的吸附性能。随着MB溶液浓度的增大,一定时间内的吸附量呈线性增加,当溶液浓度达到25 mg/L之后,增大溶液的浓度,吸附量的增量反而减少,说明0.03 g CM1∶3-2.5样品对25 mg/L的MB溶液吸附10 min后,MoS2微球表面的多孔隙结构吸附了大量的MB分子,继续增大溶液浓度,将达到吸附阈值。图7(b)可以看出,样品对不同浓度的MB溶液的吸附速率均在2 min时达到峰值,其后随时间的增加而减少。随着MB溶液浓度的增加,其峰值吸附速率也在不断增加,当浓度达到25 mg/L之后,继续增大溶液浓度,其峰值吸附速率已基本不变。这些结果表明,随着浓度的增加,MB分子与MoS2分子的接触几率越大,导致吸附速率越快[18-19]。一段时间后,MoS2微球的吸附达到饱和,剩余的吸附空位减少,吸附速率不再增大。该结果与WU[17]等对于介孔吸附的报道一致。
3.3 MoS2投放量对于吸附量的影响
选择25 mg/L的MB溶液,分别加入0.01、0.02、0.03、0.04 g CM1∶3-2.5和CM0样品,每隔2 min进行吸光度测试,总吸附时间10 min,结果如图8。
从图8(a)中可以看出,对于25 mg/L的MB溶液,在前六分钟随着CM1∶3-2.5样品投放量的增加,吸附量也不断增加,后逐步居于平稳达到吸附平衡。当CM1∶3-2.5投放量达到0.03 g时,吸附平衡后溶液基本变为无色。继续增加投放量,单位MoS2的吸附量开始下降,吸附剂过剩,曲线最大值下降。对于25 mg/L的MB溶液,当CM1∶3-2.5投放量为0.03 g时吸附效果最佳,达到38.15 mg/g。图8(b)CM0的吸附曲线表明,0.04 g CM0的吸附容量仅为32.42 mg/g,且在8 min左右达到吸附平衡,在吸附容量以及吸附速率方面都略低于CM1∶3-2.5。由于微球结构的比表面积更大于团簇状纳米片的比表面积,相同时间内MoS2微球吸附量大于团簇状纳米片MoS2的吸附量,吸附速度大大提高[20-21]。
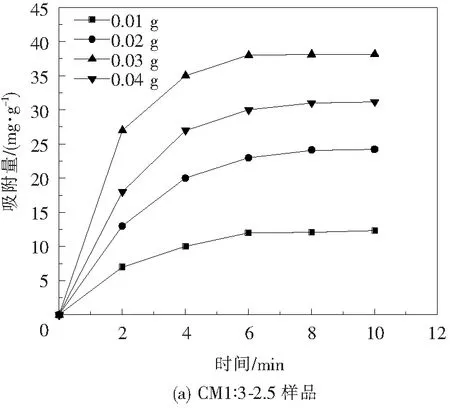
图8 MoS2投放量对单位MoS2吸附量的影响
4 总 结
用微波辅助水热法制备了自组装微球状MoS2,并解释了CTAB作为生长软模版形成微球状MoS2的自组装机制。研究了改变反应物钼硫比、生长时间对制备得到的MoS2微球形貌以及结晶度的影响。当反应时间为2.5 h,反应温度210 ℃,钼硫比为1∶3时,制备得到了分散性良好,尺寸适中且结晶度良好的MoS2微球。MoS2微球直径在1.5 μm左右,对比不添加CTAB生长得到的MoS2纳米片,CTAB在微球生长过程中起到了重要作用。BET测试表明MoS2微球比表面积为32.76 m2/g,大于MoS2纳米片,较大比表面积以及良好的介孔结构为其提供了良好的吸附性能。对MB溶液吸附测试结果表明,0.03 g MoS2微球对25 mg/L MB溶液10 min吸附量达到了38.15 mg/g,这相比于团簇状MoS2纳米片而言,成球之后吸附能力大大提升。相比于目前主流活性炭物理吸附而言,MoS2微球不但具有同样甚至更高的吸附效果而且成本更低,更因其拥有光催化降解等性能,实现可循换利用,将有望成为新一代的快速可循换吸附剂。
