椎管内富于细胞性神经鞘瘤30例临床病理分析
2021-10-12李玉洁陈晶晶吴海波
李玉洁,陈晶晶,吴海波
富于细胞性神经鞘瘤是一种少见的神经鞘瘤亚型,属于良性周围神经性肿瘤,占周围神经鞘瘤的2.8%~5.2%[1]。由于其细胞密度高,核分裂象常见,可呈局部破坏性生长,易被误诊为恶性外周神经鞘膜瘤(malignant peripheral nerve sheath tumor, MPNST)等恶性肿瘤。该肿瘤以中年人多见,最常见的受累部位是后纵隔及腹膜后脊柱旁,发生于椎管内相对少见。本文收集30例椎管内富于细胞性神经鞘瘤,探讨其临床病理学特征、免疫表型、诊断及预后,并复习相关文献,旨在提高对该肿瘤的认识。
1 材料与方法
1.1 材料收集2015年7月~2019年12月中国科学技术大学附属第一医院(安徽省立医院)确诊的椎管内神经鞘瘤409例,其中富于细胞性神经鞘瘤30例,经典型神经鞘瘤379例。回顾379例经典型神经鞘瘤的病理诊断,其中281例已行S-100免疫组化染色,215例已行SOX-10免疫组化染色。另收集7例不同部位的MPNST作为对照。
1.2 方法标本均经10%中性福尔马林固定,常规脱水,石蜡包埋,3 μm厚连续切片,分别行HE和免疫组化染色。免疫组化染色采用EnVision两步法,一抗包括SOX-10(EP268)、S-100(15E2E2)、H3K27me3(RM175),均购自北京中杉金桥公司。
1.3 结果判读H3K27me3阳性为细胞核着色,按照文献报道的评判标准[2]对着色的肿瘤细胞进行百分比评估,<5%为完全缺失,≥5%、<95%为部分缺失,≥95%为无缺失。S-100阳性为细胞核/质着色,SOX-10为细胞核着色。
1.4 统计学分析所有数据应用SPSS 21.0软件进行统计学分析,计数资料采用χ2检验比较两组之间的差异,以P<0.05作为检验水准。
2 结果
2.1 临床特征30例富于细胞性神经鞘瘤中,女性21例,男性9例,年龄20~70岁,平均52岁。其中25例位于髓外硬膜下,2例位于硬膜外,3例位于硬膜内、外。最常见的发病部位是腰椎(14例,46.7%),其次是同时累及胸椎和腰椎(10例,33.3%),胸椎(2例,6.7%),颈椎(2例,6.7%),同时累及颈椎和胸椎(1例,3.3%),同时累及腰椎和骶椎(1例,3.3%)。病灶长径0.7~8.9 cm,平均3.7 cm。临床常表现为神经压迫症状,26例出现神经支配区域明显的疼痛、麻木感,2例表现为下肢无力,1例位于椎管内圆锥处表现为小便失禁、大便困难,1例位于颈段以头晕为首发症状。379例经典型神经鞘瘤中,男性205例,女性174例,年龄4~84岁,平均51岁。亦好发生于腰椎(171例,45.1%),其次是颈椎(95例,25.1%),胸椎(73例,19.3%),同时累及腰椎和胸椎(19例,5.0%),骶椎(10例,2.6%),同时累及腰椎和骶椎(10例,2.6%),同时累及颈椎和胸椎(1例,0.3%)。临床主要表现为神经支配区域的疼痛和麻木感。7例MPNST中,女性4例,男性3例,年龄16~73岁,平均52岁。发病部位:四肢4例(57.1%),纵隔1例(14.3%),胸壁及腹壁1例(14.3%),胸椎1例(14.3%)。
2.2 影像学特征MRI显示椎间孔处结节状或类圆形软组织肿块影,5例为多发病灶,25例为单发病灶。病灶信号欠均匀,T1加权像(T1WI)呈等信号或低信号,T2加权像(T2WI)呈等信号、高信号或混杂信号,增强扫描后显示中度或明显强化(图1、2)。病灶边界尚清晰,形态规则,邻近椎管内受压。病灶沿椎间孔向外生长时,可呈现典型的哑铃状改变。

图1 腰椎MR、T1平扫示:胸12/腰1水平椎间孔扩大,其内见不规则囊实性低信号影 图2 腰椎MR、T2平扫示:胸12/腰1水平高低混杂信号影
2.3 病理检查富于细胞性神经鞘瘤常被覆纤维性包膜,本组14例可见包膜下淋巴细胞聚集灶,未见淋巴滤泡形成。瘤细胞密度较大,主要呈交织的束状排列,部分区域呈席纹状或漩涡状排列(Antoni A区)(图3)。肿瘤以Antoni A区为主,19例完全缺乏网状区(Antoni B区),11例有小灶性Antoni B区。高倍镜下,瘤细胞胞质丰富红染,境界不清,核呈梭形,染色质较粗糙,核仁不明显。部分细胞轻~中度异型性,甚至可见小核仁。多数肿瘤均可见核分裂象(1~7个/10 HPF)(图4),但无凝固性坏死。5例见散在分布的核大、深染,呈退行性改变的畸形瘤细胞,1例瘤细胞广泛退行性变。肿瘤间质血管丰富,10例见玻璃样变性的厚壁血管,17例有血管周围淋巴细胞浸润,形成血管周围淋巴套。30例均未见明显泡沫细胞沉积。肿瘤常伴出血、囊性变及含铁血黄素沉积,1例见黏液样变性,1例见骨化,2例局部有钙化。
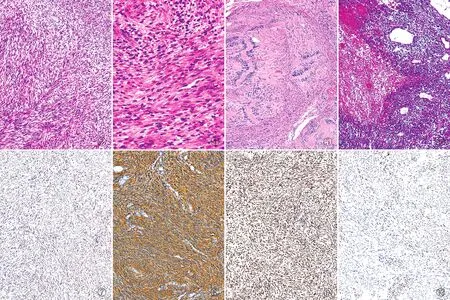
图3 富于细胞性神经鞘瘤,呈交织束状排列 图4 富于细胞性神经鞘瘤,瘤细胞丰富,易见核分裂象 图5 经典型神经鞘瘤,易见栅栏状排列和Verocay小体 图6 恶性外围神经鞘膜瘤,瘤细胞异型性明显,可见地图状坏死 图7 富于细胞性神经鞘瘤,瘤细胞SOX-10弥漫阳性,EnVision法 图8 富于细胞性神经鞘瘤,瘤细胞S-100弥漫阳性,EnVision法 图9 富于细胞性神经鞘瘤,瘤细胞H3K27me3弥漫阳性,EnVision法 图10 富于细胞性神经鞘瘤,瘤细胞H3K27me3部分缺失,EnVision法
经典型神经鞘瘤均由Antoni A区和Antoni B区交替分布组成,常见栅栏状排列,Verocay小体(图5)及包膜下淋巴细胞聚集灶。瘤细胞无明显异型,核仁不明显,核分裂象不易见。间质可见玻璃样变性的厚壁血管,常伴出血、囊性变及含铁血黄素沉积,易见片状泡沫细胞沉积。MPNST由短梭形细胞、卵圆形细胞呈束状、交织状或漩涡状排列,局部似纤维肉瘤样(鱼骨样或人字形排列),1例周围见神经纤维瘤样区域。瘤细胞密度较大,呈弥漫浸润性生长。5例见纤维性假包膜,其中1例见包膜下淋巴细胞聚集灶。瘤细胞弥漫性中~重度异型,染色质粗块状,核仁不明显。部分核呈空泡状,明显的小核仁。核分裂象多见(6~53个/10 HPF),可见非典型性核分裂象,散在奇异形瘤巨细胞,1例伴横纹肌样分化。肿瘤间质血管丰富,1例见玻璃样变性的厚壁血管,1例见血管周围淋巴套。6例见地图状坏死(图6)。
2.4 免疫表型富于细胞性神经鞘瘤表达至少一种施万细胞标志物,包括SOX-10核阳性(30/30)(图7),S-100核和质弥漫阳性(28/30)(图8)。14例H3K27me3弥漫阳性(图9);16例H3K27me3部分缺失(图10),但肿瘤细胞失表达率均﹤50%。379例经典型神经鞘瘤中,随机抽取50例行H3K27me3免疫组化染色,19例无缺失,31例部分缺失,肿瘤细胞失表达率均﹤50%。H3K27me3在经典型神经鞘瘤和富于细胞性神经鞘瘤中的表达差异无统计学意义(P>0.05)。既往行S-100和SOX-10免疫组化的病例,1例S-100阴性,1例SOX-10阴性,其余病例两者均阳性。7例MPNST中,有2例H3K27me3完全缺失,同时S-100和SOX-10阴性;其余5例H3K27me3部分缺失,其中3例肿瘤细胞失表达率>50%,2例失表达率<50%,S-100和SOX-10均灶阳性。
2.5 随访30例富于细胞性神经鞘瘤患者术后均未接受放、化疗,4例失访,26例电话随访3~55个月(平均24个月),所有患者均健在,未发生复发或转移。本组中1例神经鞘瘤于2005年行肿块切除术,2019年相同部位复发,病理诊断为富于细胞性神经鞘瘤,随访至今,未发现再次复发或转移。
3 讨论
富于细胞性神经鞘瘤于1981年由Woodruff等[3]首次提出,是一种少见的特殊类型神经鞘瘤,与经典型神经鞘瘤相似,可发生于人体的各个部位,尤其好发于后纵隔和腹膜后脊柱旁,其以高密度、轻~中度的核异型性和较易见的核分裂象引起关注。该肿瘤易误诊为各种类型的梭形细胞肉瘤,尤其是MPNST。椎管内的富于细胞性神经鞘瘤相比其他部位更为少见,其好发于中年人,女性多见[4],本组女性21例,男性9例,平均年龄52岁。文献报道椎管内神经鞘瘤最常见于髓外硬膜下,可出现在脊椎的任何部位,好发于腰椎和颈椎[5]。本组椎管内富于细胞性神经鞘瘤以髓外硬膜下最多见,腰椎和胸椎好发,与文献报道基本一致。影像学上,MRI常显示病灶边界较清楚。当肿瘤沿椎间孔向外生长时,可呈现典型的哑铃状改变。T1加权像(T1WI)呈等信号或低信号,T2加权像(T2WI)呈等信号、高信号或混杂信号,增强扫描后显示中度或明显强化,如出现囊性变则显示为低信号。
组织学上,富于细胞性神经鞘瘤的梭形细胞呈紧密的交织束状排列,缺乏或仅有局灶的Antoni B区(小于肿瘤面积的10%)[6]。本组30例椎管内富于细胞性神经鞘瘤表现为相对较一致的形态学特征。瘤细胞常有轻~中度异型性,可出现核仁,核分裂象最高达7个/10 HPF,这些特征易误诊为MPNST(表1)。除了出现经典型神经鞘瘤常见的局部压迫,富于细胞性神经鞘瘤有时还可侵犯周围骨组织导致溶骨性破坏,但这并不是恶性依据[7]。其生长缓慢,肿瘤常出现陈旧性出血、囊性变、血管壁玻璃样变性等退行性改变,此外还可见核大、深染的退变瘤细胞,但钙化和骨化较少见。Din等[8]的报道显示约1.8%神经鞘瘤出现钙化和骨化,钙化包括营养不良性钙化和砂粒体型钙化,主要出现在Antoni A区,与Verocay小体密切相关,而骨化主要出现在Antoni B区。本组有2例钙化,1例骨化,均见于Antoni A区,2例钙化均为营养不良性钙化。大多数富于细胞性神经鞘瘤仍可见经典型神经鞘瘤的形态学特征,如包膜下淋巴细胞聚集灶、血管周围淋巴套、含铁血黄素沉积等,这些特征通常仅限于局部,需仔细观察,有助于作出正确诊断。
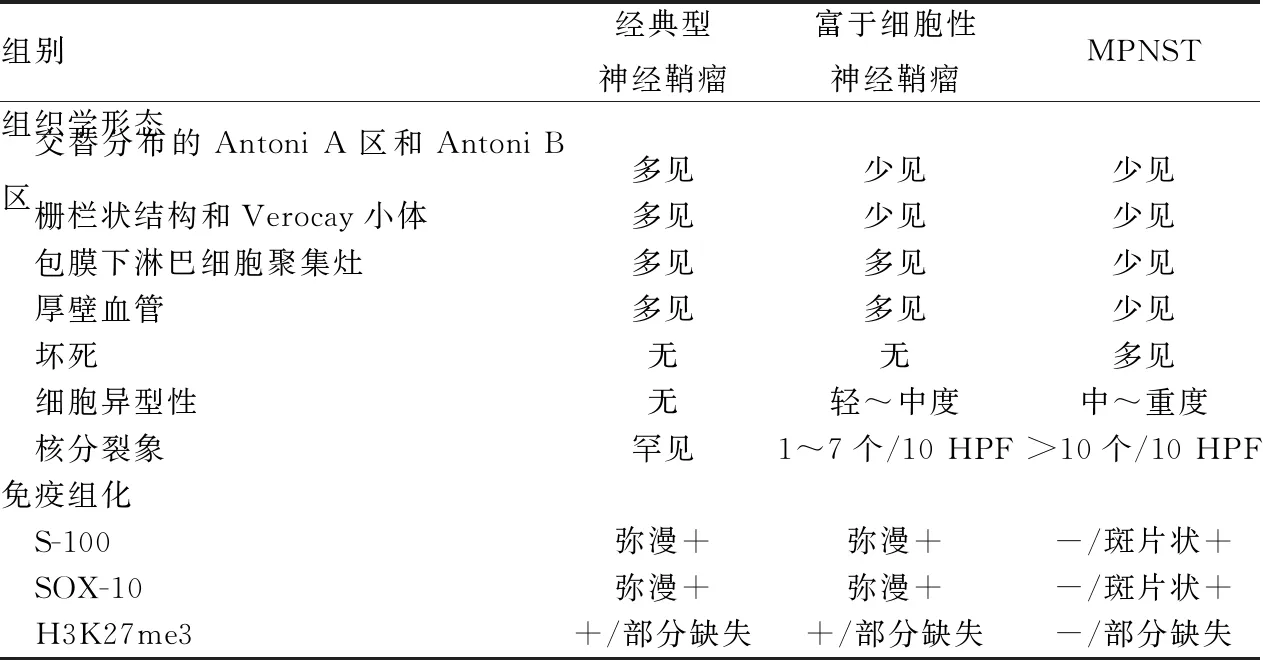
表1 富于细胞性神经鞘瘤与经典型神经鞘瘤、MPNST的鉴别诊断
经典型神经鞘瘤与富于细胞性神经鞘瘤的区别为:有交替分布的Antoni A区和Antoni B区,易见栅栏状结构和Verocay小体,瘤细胞密度不高,无明显异型性,核分裂象罕见。两者免疫表型较一致,至少表达一种施万细胞标志物(S-100或SOX-10),H3K27me3无缺失或部分缺失(肿瘤细胞失表达率均﹤50%),两者H3K27me3表达差异无统计学意义(P>0.05),因此仅凭免疫组化并不能很好的区分两者,主要依赖于组织形态学特征。
富于细胞性神经鞘瘤最重要的鉴别诊断是MPNST,两者的治疗方法和预后均不同。富于细胞性神经鞘瘤只需手术完全切除即可,而MPSNT术后需要辅助放疗和化疗[9-10]。MPNST恶性度较高,易复发和转移,预后较差,5年生存率为32%~50%[11-13],而富于细胞性神经鞘瘤的5年生存率为100%[14]。MPNST多数直接起源于神经纤维瘤,约50%伴Ⅰ型神经纤维瘤病,瘤细胞有明显异型性,核分裂象多见(常>10个/10 HPF),地图状坏死常见。此外,少数MPNST伴有异源性分化,如横纹肌、软骨和骨等[15-16],而富于细胞性神经鞘瘤罕见异源性分化。免疫组化可以辅助鉴别诊断,目前最常用的周围神经标志物S-100和SOX-10在MPNST中的阳性率分别为40%~50%、30%[17],常为斑片状阳性,而在富于细胞性神经鞘瘤中几乎均弥漫阳性。既往研究表明[17-18],大多数MPNST存在多梳抑制复合物2(PRC2)的失活,导致H3K27me3表达缺失。EED和SUZ12的基因表达产物是构成PRC2复合物核心的重要成分,主要负责组蛋白3的赖氨酸27位点的甲基化(包括三甲基化和二甲基化),PRC2失活是由于SUZ12或EED1的失活突变引起,因此认为H3K27me3缺失可以作为MPNST的诊断指标。需要注意的是,H3K27me3的敏感性和特异性并不高,其表达缺失在MPNST中的发生率仅为34%~51%,在滑膜肉瘤、低级别纤维黏液样肉瘤、梭形细胞黑色素瘤、梭形细胞横纹肌肉瘤、隆突性皮肤纤维肉瘤等肿瘤中均存在H3K27me3部分缺失[2,17-19]。有学者认为H3K27me3表达缺失更常见于高级别MPNST,对于低级别和中等分化MPNST并不是敏感的标记[20]。本组发现在经典型神经鞘瘤和富于细胞性神经鞘瘤中也可出现H3K27me3部分缺失,但均﹤50%。这提示H3K27me3部分缺失不能区分富于细胞性神经鞘瘤和MPNST,但当H3K27me3部分缺失>50%时,对MPNST的诊断有一定提示作用。而Asano等[2]认为H3K27me3完全缺失(<5%肿瘤细胞着色)才具有诊断的特异性。Wu等[21]发现染色质域解旋酶DNA结合蛋白4(CHD4)在两者中的表达位置不同,富于细胞性神经鞘瘤细胞胞质和胞核均有表达,但在MPNST中仅细胞核表达,有助于鉴别两者。
此外,椎管内富于细胞性神经鞘瘤还需与以下病变相鉴别,(1)椎管内室管膜瘤:细胞也可呈交叉的束状排列,并表达S-100和GFAP,但室管膜瘤常有特征性的血管周围假菊形团和室管膜真菊形团,EMA环状或核旁点状阳性,SOX10常不表达,均有助于鉴别。(2)纤维型脊膜瘤:瘤细胞梭形,呈交织状、束状或漩涡状排列,与富于细胞性神经鞘瘤相似,但纤维型脊膜瘤表达EMA、SSTR2α和PR,不表达S-100和SOX-10,可资鉴别。(3)孤立性纤维性肿瘤(solitary fibrous tumor, SFT):常由交替分布的细胞疏松区和致密区组成,易见鹿角状血管,免疫组化标记STAT-6、CD34、CD99和BCL-2阳性,S-100和SOX-10阴性。(4)梭形细胞型滑膜肉瘤:瘤细胞梭形,呈交织状、束状排列,可表达S-100蛋白,存在H3K27me3部分缺失,但可表达TLE1、EMA、CK,大多数存在染色体易位t(X;18)(p11.2;q11.2)及SYT-SSX融合基因。
富于细胞性神经鞘瘤属于良性肿瘤,行手术完整切除后一般预后良好,不再复发。既往报道显示,富于细胞性神经鞘瘤患者的术后5年无进展生存率和5年无瘤生存率均为100%[14]。对于手术切除不完全的肿瘤,会局部复发,但未发现远处转移[22]。本组病例术后均未行放疗和化疗,所有患者预后良好,未发生复发和转移。其中1例神经鞘瘤复发后转变成富于细胞性神经鞘瘤,复发的原因可能与初次手术未完全切除有关,该例患者随访至今未出现再次复发。
综上所述,椎管内富于细胞神经鞘瘤较少见,与MPNST等肿瘤鉴别较困难。通过仔细观察其形态学特征,并结合免疫组化综合判断,有助于诊断和鉴别诊断。由于该肿瘤有复发风险,建议对患者进行长期随访。
