基于ESPCA模型的结肠癌差异基因筛选研究*
2021-10-09哈尔滨医科大学卫生统计学教研室150081张秋菊丛雨欣刘美娜
哈尔滨医科大学卫生统计学教研室(150081) 李 称 张秋菊 孙 琳 丛雨欣 赵 敏 刘美娜
【提 要】 目的 验证ESPCA模型降维及变量选择的效果优于SPCA;利用ESPCA模型分析结肠癌基因数据,寻找不同主成分的通路信息,筛选结肠癌的差异基因。方法 模拟实验比较ESPCA模型与SPCA的降维及变量选择能力,通过灵敏度、特异度、准确度评价其变量筛选效果;收集TCGA结肠癌基因数据,利用ESPCA模型对结肠癌基因数据进行降维及变量筛选。结果 模拟实验获得的ESPCA模型具有较高的灵敏度、特异度和准确度,降维及变量选择效果优于SPCA。结肠癌数据分析结果显示:降维后ESPCA模型第一主成分中基因与18条GO-BP通路及7条KEGG通路有关,ESPCA模型第二主成分中基因与19条GO-BP通路有关;根据度中心性及中介中心性对主成分基因排序筛选出6个差异基因:MYC、CD44、PKM、FBL、PSMA7、RPS2,经GSE137327数据集验证具有较高的AUC值。结论 ESPCA模型在降维过程中既考虑数据本身信息,又考虑了生物网络信息,具有良好的降维及变量选择效果。结肠癌数据分析中ESPCA第一主成分中基因参与癌症相关的通路;ESPCA第二主成分中基因参与的通路与免疫相关;通路中基因参与癌症的发生发展过程、可能通过免疫反应相关通路调节结肠癌的发生发展过程;获得的6个结肠癌差异基因可为研究疾病发生机制和鉴别诊断提供依据。
随着组学技术发展,高通量组学数据为寻找生物标志物、疾病鉴别诊断提供新机遇。“高维、小样本”为组学数据的重要特征,成为影响组学研究结果准确性的重要因素。为解决这一问题,许多数据降维、变量选择方法被应用于组学数据分析。目前,稀疏主成分分析(sparse principal component analysis,SPCA)广泛用于高维组学数据降维,如LASSO正则化[1]、加权稀疏主成分分析[2]。SPCA模型在基因筛选中,假定所有基因有同等概率被选至各主成分中,没有考虑变量间的权重、忽略基因在网络中的相互作用。边组稀疏主成分分析[3](edge-group sparse PCA,ESPCA)以基因调控网络中相互作用关系为先验信息对高维组学数据进行分析;基因调控网络[4]可以反应出基因间相互作用关系,将调控网络引入到稀疏主成分分析中,获得的主成分更加接近真实的生物模式,但该方法尚未应用在癌症基因数据的变量筛选中。
目前,结肠癌的发病率逐年递增,基因治疗[5]正被广泛用于肿瘤等疾病的治疗中,但其潜在的基因通路及结肠癌差异基因仍不明确。本研究通过模拟比较ESPCA模型与SPCA在降维、变量选择的效果,并利用ESPCA模型对结肠癌基因数据集进行降维及变量选择,获得结肠癌相关通路及差异基因,为研究结肠癌的相关机制及鉴别诊断与治疗提供依据。
分析方法
1.ESPCA 模型
ESPCA模型[3]是一种以网络交互作用为导向处理高维组学数据的方法,将稀疏主成分与先验网络相结合,通过选择具有交互作用的组结构实现变量选择,算法步骤为:
(1)假设g为组结构,g={e1,…,eM}表示基因调控网络中所有边组的集合。当g为非重叠的组结构时,组稀疏惩罚为L1范数或L0范数。然而实际应用中需要考虑重叠组结构[6]的情况,基因相互作用网络中两个相连基因被视为一个边组。显然,边组为重叠组结构,因此边组稀疏惩罚(ES惩罚)如下:
‖u‖ES=minimize|g′|
∀g′⊆g,support(u)⊆V(g′)
(1)
其中,g′为g的子集,V(g′)为g′的顶点集,|g′|表示g′的元素个数,support(u)为u中非零元素指数集。
(2)由于ES惩罚的应用,ESPCA载荷非零元素的选择基于g中重要基因相互作用的边。最终形成带有ES惩罚的主成分分析即ESPCA,其表达式如下:
(2)
其中,X为基因表达矩阵,u为主成分载荷,v为主成分,k为选择边的个数,‖·‖2为欧氏距离。
(3)模型参数估计的核心问题在于用固定的v、z=Xv,求解k边组稀疏投影问题。该问题为NP-难问题,将通过贪婪算法[7]求解下式的最优解:
(3)
(4)
其中,g(i)为包含基因i的边组指数集;supp(z,k)表示z的绝对值中较大的k个元素的指数集。
(4)采用交替迭代策略直到收敛得到最优解:
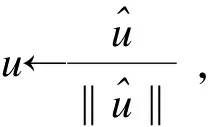
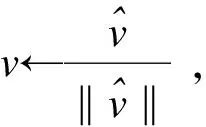
(5)应用Gram-Schmidt正交化方法,分别求出ESPCA多个主成分和主成分载荷。
(6)
2.模型参数选择
边个数k和主成分数l是ESPCA模型最重要的参数,这两个参数的确定是开放性的问题,常以最小化贝叶斯信息准则(bayes′ information criterion,BIC)实现模型参数估计为可行的策略之一。根据已有文献[3],本研究将k=150,l=2设为默认值。
模拟实验
1.模拟参数设置
(1)生成两个主成分载荷u1、u2,长度为200的列向量,u1中前100个变量的载荷随机产生,后100个变量载荷均为0;u2反之;
(2)生成两个主成分v1、v2,表示随机产生的长度为100的列向量;
(4)相互作用网络g由g1,g2,g3组成;g1,g2分别为前100和后100个变量以概率p=0.3组成的网络;g3为前、后100个变量之间以p=0.04组成的网络;
(5)k设定为300,为了便于比较,提取前两个主成分,且SPCA每个主成分提取变量个数与ESPCA的相同。
2.模拟评价指标
(1)灵敏度:实际为阳性(系数非零)的变量中,判断为阳性的比例。
(2)特异度:实际为阴性(系数为零)的变量中,判断为阴性的比例。
(3)准确度:正确判断为阳性和阴性变量的比例。
3.模拟实验结果
模拟实验获得两个主成分,每一个主成分在不同的噪声水平下,ESPCA灵敏度、特异度、准确度的效果优于SPCA;随着噪声水平γ的增大,灵敏度、特异度、准确度呈下降趋势;由于PC1大于PC2的权重值,故PC1有更高的灵敏度、特异度、准确度,见图1。
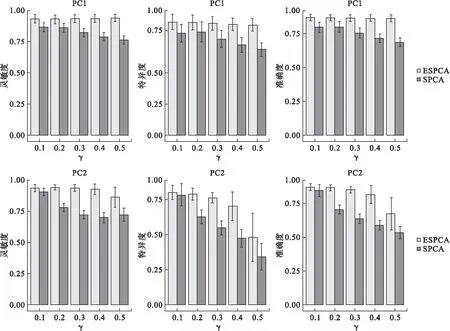
图1 在不同噪声水平下SPCA、ESPCA的灵敏度、特异度、准确度
结肠癌差异基因筛选
下载TCGA(TCGA,http://cancergenome.nih.gov/)结肠癌RNA-seq表达数据:包括60488个基因,512例样本(471个为癌组织,41个为癌旁组织)。
1.数据预处理
基因筛选:(1)根据“80原则”剔除缺失变量;(2)相同探针保留变异较大的基因;(3)剔除t检验P>0.05的变量,最终剩余7837个基因。边的获得:利用7837个基因在STRING网站中获得基因网络相互作用的边,共87493条。
2.ESPCA模型降维及富集分析
ESPCA模型应用于预处理后结肠癌数据集,提取前两个主成分:ESPCA1和ESPCA2,绘制结肠癌得分图,结果见图2,图中两个主成分可以很好地区分癌组织与癌旁组织。
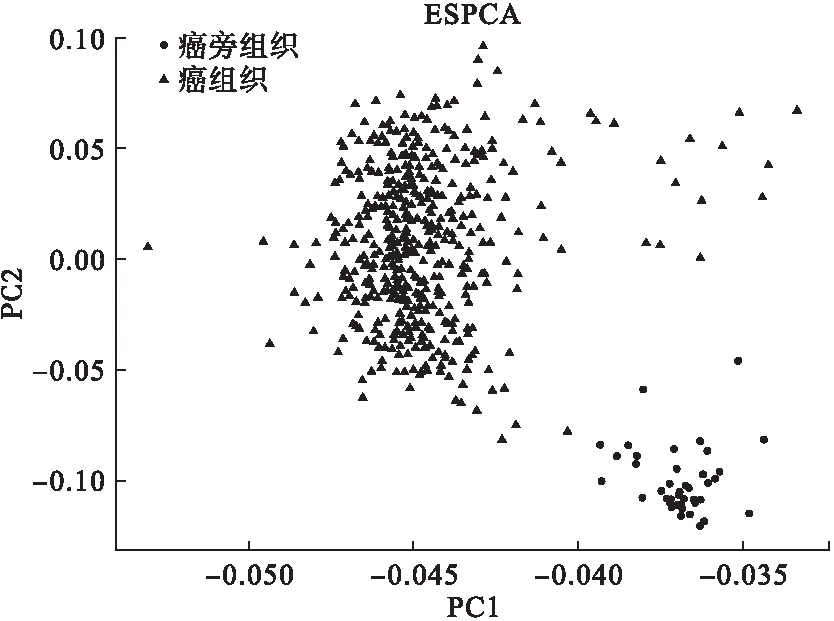
图2 基于ESPCA前两个主成分结肠癌的得分图
ESPCA1中有95个基因变量,ESPCA2中有92个基因变量,将两个主成分中的基因分别进行富集分析发现:ESPCA1中基因与18条GO-BP通路和7条KEGG通路有关,其中细胞分裂、PI3K-Akt信号、EMC-受体交互、癌症中心的碳代谢等通路与癌症的发生发展相关,如 PKM,SLC1A5,SLC2A1,MYC等基因参与KEGG中的癌症中心碳代谢通路,结果见表1;ESPCA2的基因与19条GO-BP通路有关,富集分析发现通路与免疫过程相关,如:补体激活、免疫反应调节、B细胞受体信号通路、免疫应答等,结果见表2。
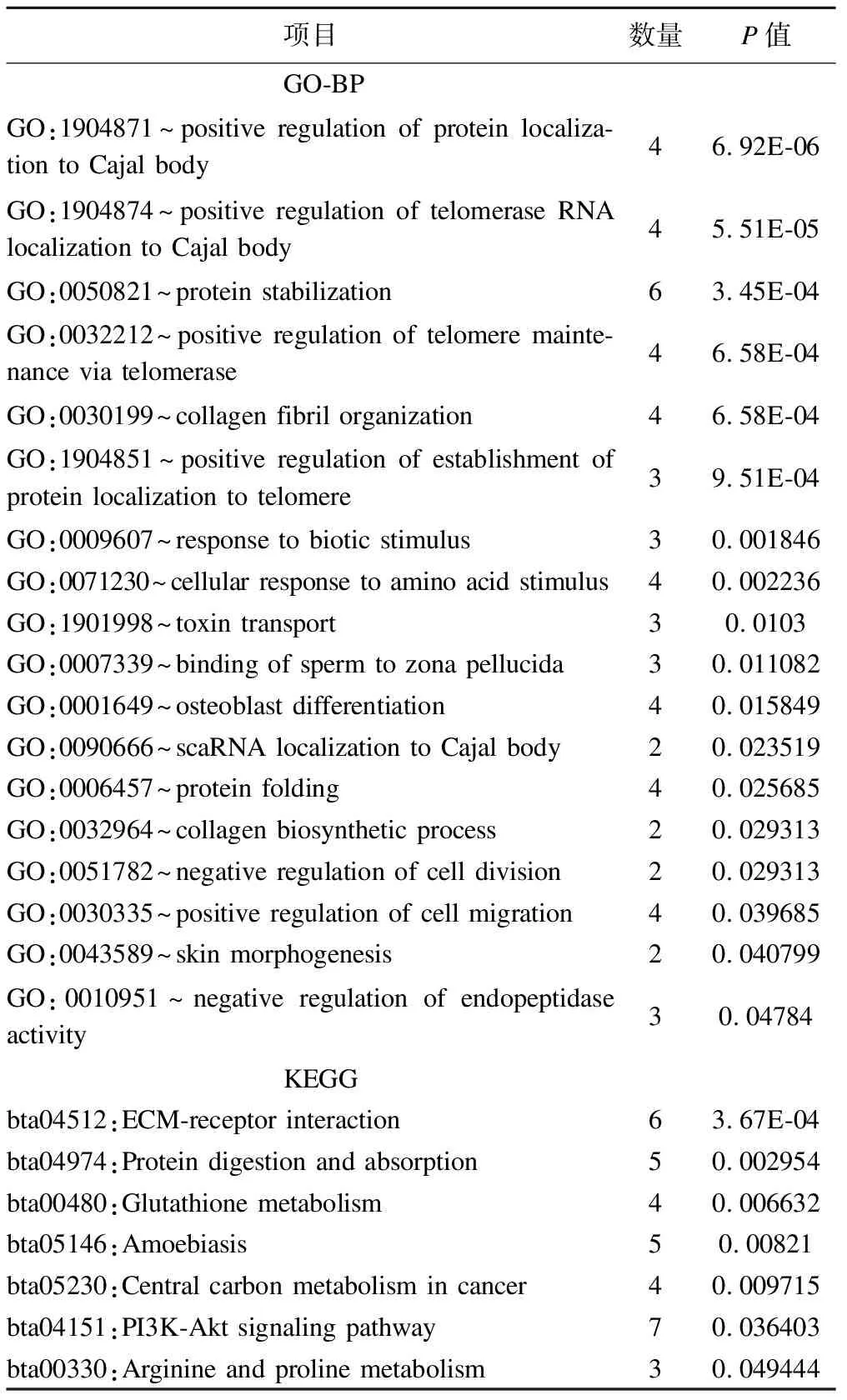
表1 ESPCA第一主成分富集分析结果
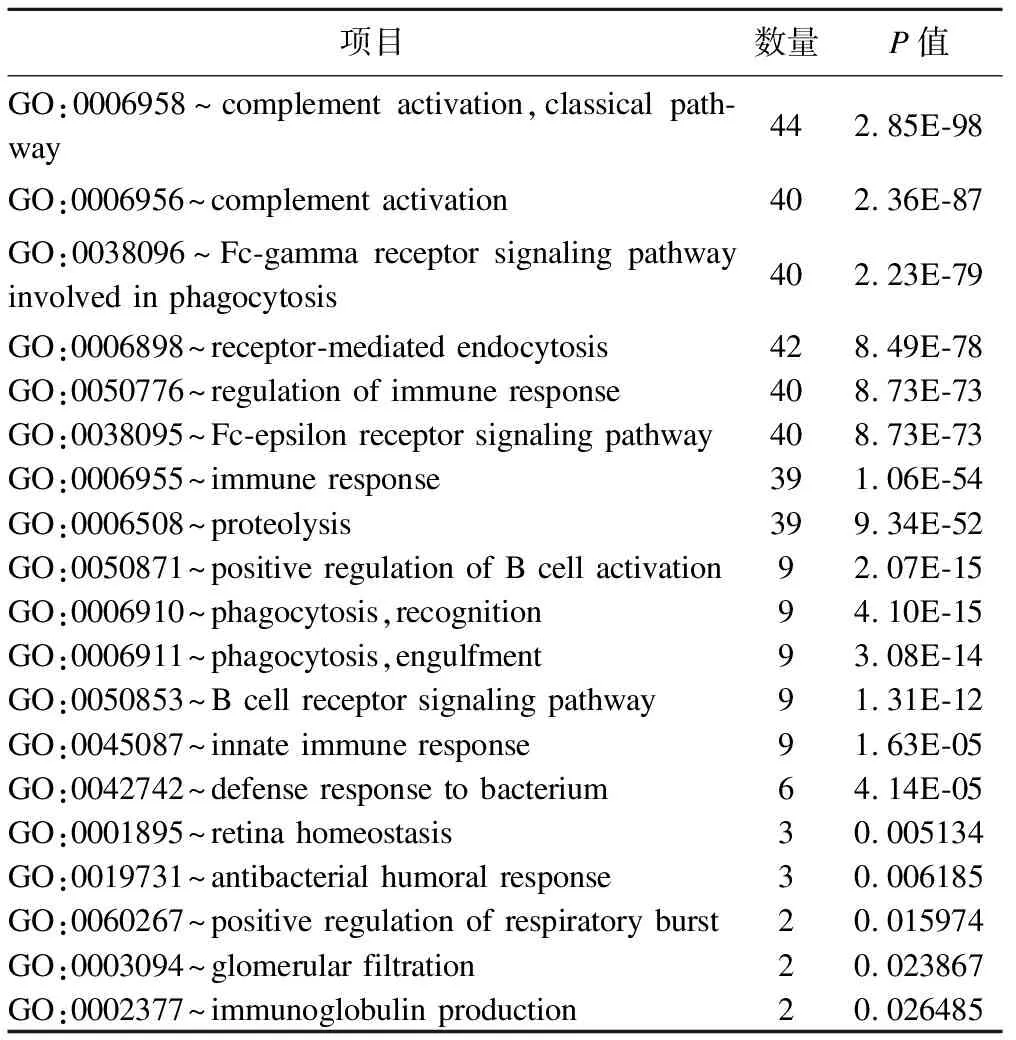
表2 ESPCA第二主成分富集分析结果
3.筛选结肠癌的差异基因
主成分中的基因做PPI网络图(图3),根据度中心性和中介中心性排序,选择前6个基因,分别为MYC、CD44、PKM、FBL、PSMA7、RPS2,其AUC值分别为0.97、0.99、0.98、0.96、0.97、0.96,见表3。
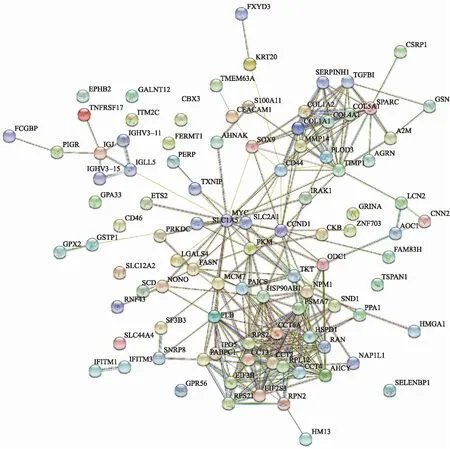
图3 基因相互作用网络
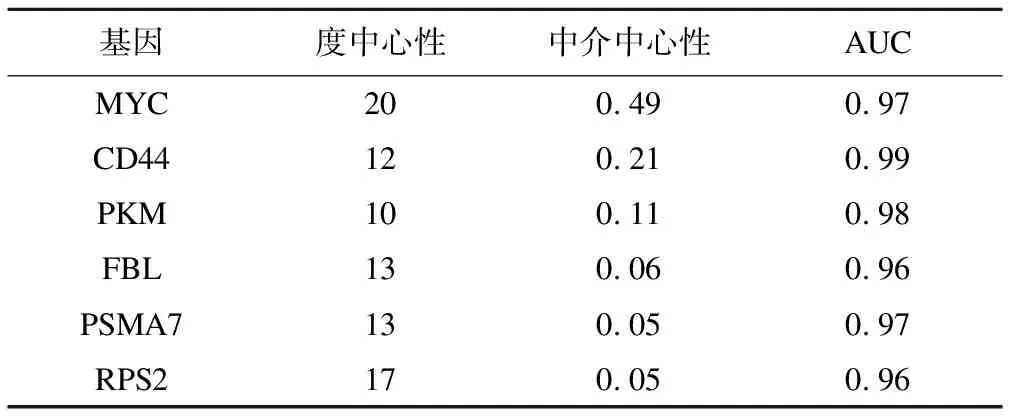
表3 6个基因排序及AUC值
下载GEO结肠癌数据集GSE137327共18例样本,其中9个为对照组,9个为病例组进行外部验证,6个基因的AUC值分别为0.72、0.69、0.74、0.77、0.68、0.74,结果见图4。
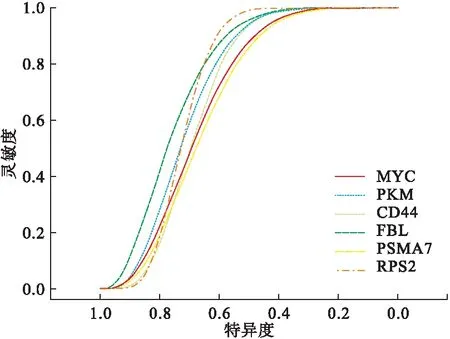
图4 筛选6个基因的ROC曲线
讨 论
本研究模拟实验的验证结果显示,在不同的噪声水平下,ESPCA的灵敏度、特异度、准确度均优于SPCA,在边的信息下提取的样本信息更加接近真实模式。ESPCA模型不仅能用于基因组学数据,也可用于对蛋白质组学、代谢组学数据的降维及变量选择。
ESPCA模型对结肠癌数据进行降维分析,获得两个主成分ESPCA1和ESPCA2,基于两个主成分可以很好地区分癌组织和癌旁组织。富集分析发现ESPCA1中的基因参与PI3K-Akt信号、EMC-受体交互、癌症中心的碳代谢等与癌症相关的通路。研究表明:PI3K-Akt信号通路[8]是癌症中影响生存进展的主要通路,Akt[9]的失调会导致癌症、糖尿病、心血管和神经系统等疾病;EMC[10]对细胞粘附、增殖、凋亡起重要作用,影响肿瘤的进展过程;与正常细胞相比,癌症细胞的中心代谢通路[11]存在明显差异,癌症细胞即使在正常的氧气浓度下也能将大部分葡萄糖转换为乳酸,这种差异影响癌症的发生发展。富集分析发现ESPCA2中的基因参与补体激活、免疫反应调节、B细胞受体信号、免疫应答等免疫相关通路。有研究发现,免疫浸润有关的mRNA与I~III期的结肠癌的诊断及预后有关;肿瘤浸润淋巴细胞是三阴性乳腺癌[12]重要的预后因素。目前尚未发现免疫通路影响结肠癌的发生,提示基因通过调控免疫反应通路可能影响结肠癌的发生发展,免疫反应机制可能是发生癌症的潜在机制之一。
筛选出的6个基因在结肠癌数据集及GSE137327验证集中有较高的AUC值。其中,PKM、MYC、CD44和PSMA7已被证实与结肠癌有关,PKM[13]基因通过调控STAT3相关的信号通路促进结肠癌细胞的黏附和迁移;MYC[14]基因为结肠癌的关键基因,并作为LEF1的转录调节因子,通过激活LEF1的表达来调节结肠癌细胞的增殖过程;CD44[15]基因已被多个研究证实是结肠癌和胃癌干细胞的生物标志物;PSMA7[16]基因在结肠癌组织中过表达,能诱导HT-29细胞的凋亡。目前尚未发现FBL、RPS2与结肠癌发生发展有关,本研究为进一步深入了解结肠癌发生发展过程及基因治疗提供了参考和依据。
本文虽然扩展了现有文献的参数设置,但仍需探索更多的参数组合下模型的效果;GSE137327验证集中癌组织和癌旁组织的比例与TCGA结肠癌数据集中的比例相差较大,结果可能会有一定的偏差;只考虑了网络中两个相互作用的基因作为一个组结构,未来可以考虑将通路中具有类似功能的多个基因作为一个组结构进行ESPCA模型分析。
