番鸭呼肠孤病毒p10.8蛋白单克隆抗体的制备
2021-10-08屈贵蜀严梦涵陆芍华黄瑞玲彭瑶顺庄许诺许丽惠王全溪
屈贵蜀, 何 晴, 严梦涵, 陆芍华, 黄瑞玲, 彭瑶顺, 庄许诺, 许丽惠, 王全溪
[福建农林大学动物科学学院(蜂学学院),福建 福州 350002]
番鸭呼肠孤病毒(muscovy duck reovirus, MDRV)是一种具有较高致病性的双链RNA病毒[1],其引起的番鸭呼肠孤病毒病于1950年首次报道于南非,上世纪90年代,我国福建、广东等地也发现了此病[2-5].该病主要感染雏番鸭,番鸭感染之后表现出较高的发病率和死亡率[6].在番鸭的养殖中,该病已经是影响其养殖业的重要疾病之一.值得注意的是,MDRV感染引发的高致死率与其诱导的细胞凋亡密切相关[7].研究表明,MDRV中编码的非结构蛋白p10.8能够诱导细胞凋亡[8],且p10.8蛋白能够通过PERK/eIF2α通路诱导内质网应激,从而介导p10.8蛋白的细胞周期阻滞和凋亡[9],p10.8蛋白诱导细胞周期阻滞和凋亡还需要Cdc20及分子伴侣CCT2和CCT5的参与[10].可见,p10.8蛋白是引起MDRV致病的关键蛋白.
单克隆抗体(monoclonal antibody, mAb)技术,因其特异性强、亲和力高等多种优点,被广泛应用于兽医临床的检测与治疗[11].目前,刘思伽等[12]研制了 MDRV的弱毒疫苗;朱小丽等[13]制备了MDRV的mAb,但未确定是针对何种病毒蛋白;张云等[14]制备了抗MDRV σB蛋白的mAb;朱二鹏等[15]制备了MDRV σNS蛋白的多克隆抗体;吕小婷等[4]制备了MDRV σA蛋白的多克隆抗体;庄育彬[16]制备了MDRV σC蛋白的多克隆抗体.本课题组前期研究表明,p10.8蛋白可以通过非经典途径进入细胞核,但其不具有核定位信号[17].本试验力求通过p10.8蛋白的mAb去捕获与p10.8蛋白相互作用的蛋白,从而深入了解p10.8蛋白进入细胞核的相关机制.目前,MDRV p10.8蛋白mAb的研究尚未见报道,且MDRV p10.8蛋白是其致病的关键蛋白,因此开展MDRV p10.8蛋白mAb的研究对于深入探究MDRV的致病机制具有十分重要的意义.
1 材料与方法
1.1 材料
高纯度质粒小提中量试剂盒(DP107)购自北京天根生化科技有限公司;6~8周龄小鼠购自福建省福州市吴氏动物贸易有限公司;大肠杆菌BL21(DE3)感受态细胞、ProteinIsoTMNi-NTA Resin蛋白纯化柱(DP101-01)、间接ELISA法所用试剂均购自北京索莱宝科技有限公司;SP2/0细胞由福建农林大学动物科学学院提供;DF1细胞、p10.8原核表达蛋白和真核表达蛋白均来自于本实验室;其余常规试剂均购自国药试剂公司.
1.2 免疫抗原的制备
参考蔡一龙[18]MDRV p10.8蛋白原核表达及蛋白纯化的方法.将本实验室保存的p10.8蛋白原核重组菌复苏,提质粒,转化至大肠杆菌BL21感受态细胞中,在最适温度为37 ℃,IPTG最适浓度为1.0 mmol·L-1的条件下诱导24 h,使目的蛋白大量表达,收集菌液离心,变性,采用SDS-PAGE电泳检测p10.8蛋白是否表达.对诱导表达后的菌液进行离心,用PBS洗两次,加蛋白平衡液进行超声破碎,最后用Ni-NTA亲和层析法进行重组蛋白纯化,用BCA蛋白浓度检测法测定蛋白浓度.
1.3 小鼠的免疫
取饲养达7周龄的健康BALB/c小鼠,用纯化的重组p10.8蛋白作为抗原,按照抗原与弗氏佐剂的体积比为1∶1进行混合,皮下多点免疫小鼠4次(每隔两周1次),抗原免疫剂量为50 μg·只-1·次-1.第1次免疫用1 mL抗原与弗氏完全佐剂的混合液(1∶1),第2、3次用抗原与弗氏不完全佐剂的混合液(1∶1),共1 mL.前3次均采用背颈皮下注射,第3次免疫后尾静脉采血,分离血清,用Western blotting法检测血清抗体效价.达到合格效价后,腹腔注射纯化后的抗原(50 μg·只-1)以加强免疫.3 d后采用拉颈的方法将小鼠处死,无菌取出脾脏,用于后续的细胞融合试验.
1.4 细胞融合与阳性杂交瘤细胞株的筛选
细胞融合的前一天,将未免疫的小鼠腹腔巨噬细胞铺在10块96孔板上作为饲养细胞.融合当天,在无菌条件下取小鼠脾脏,分离脾细胞,将脾细胞用5 mL离心管收集起来,并计数.将处于对数生长期的骨髓瘤细胞SP2/0从培养箱取出,离心,计数,按照脾细胞∶SP2/0细胞以10∶1的数量比进行混合.将PEG诱导剂和DMEM培养基分别用水浴锅(37 ℃)预热,取1 mL PEG诱导剂加入到脾细胞和骨髓瘤细胞的混合液中,由慢到快,60 s内加完.在加入的同时需要搅拌或者摇晃,加完之后再摇1.5 min.在5 min内加入10 mL DMEM培养液,然后再加40 mL DMEM培养液,离心,去上清.加入10 mL HAT培养液,按每孔100 μL的量分装于24孔板中并放入细胞培养箱中培养.融合10~15 d后,用间接ELISA法检测细胞上清效价,挑选D450 nm高的、细胞生长旺盛且形态好的、没有交叉反应的阳性孔,转移到24孔板中.第2天观察细胞生长情况,选取细胞生长状况好的进行计数.采用有限稀释法,经3次连续克隆化,直至板中各孔阳性率达100%,进行克隆化的过程中各阶段细胞需要扩大培养并收集上清.
1.5 p10.8 mAb特异性鉴定
1.5.1 mAb的Western blotting鉴定 以p10.8 mAb阳性细胞株培养的上清为一抗,分别与原核表达、真核表达的p10.8蛋白进行Western blotting反应;另取MDRV、禽腺病毒(FAdV)和鸭坦步苏病毒(DTMUV)全病毒与mAb细胞培养的上清进行反应.试验均同时设置阴性对照检测其生物学活性.
1.5.2 mAb效价及亚类的测定 取阳性克隆株细胞培养上清,用间接ELISA法检测抗体效价;按照齐一生物科技(上海)有限公司的单克隆抗体亚类检测试剂盒说明书的方法检测抗体亚类.
2 结果与分析
2.1 pET-32a-p10.8原核重组蛋白的表达及纯化
将重组质粒pET-32a-p10.8转化BL21(DE3)感受态细胞24 h,分别以p10.8基因的上下游引物、T7的上下游引物、p10.8基因的上游引物和T7的下游引物、T7的上游引物和p10.8基因的下游引物进行PCR扩增鉴定,在预期的相应位置出现特异性条带(图1A).SDS-PAGE检测结果显示,p10.8蛋白被诱导表达,其特异性条带位于预期大小的28.8 ku处(图1B),纯化效果好,杂蛋白少(图1C).

A:重组质粒转化BL21的PCR鉴定(M:2 000 bp DNA Maker;1:p10.8基因的上下游引物;2:T7的上下游引物;3:p10.8基因的上游引物、T7的下游引物;4:T7的上游引物、p10.8基因的下游引物);B:重组菌的诱导表达(M:180 ku蛋白Maker;1:上清;2:沉淀;3:pET-32a空载体表达组); C:重组菌的纯化(M:180 ku蛋白Maker;1:pET-32a空载体表达组;2:抗原的纯化).图1 p10.8蛋白原核表达及纯化的检测结果Fig.1 Prokaryotic expression and purification of p10.8 protein
2.2 小鼠阳性血清效价检测结果
Western blotting检测结果表明,3免后小鼠血清效价达1∶32 000(图2).对小鼠阳性血清进行梯度稀释至1∶256 000,采用间接ELISA法进一步检测血清的酶标值(D450 nm)以判定其效价.结果(表1)显示,效价在1∶256 000时,免疫组血清的酶标值仍达0.811 5.

1:PBS空白对照组,2:p10.8原核表达蛋白组.图2 血清效价达1∶32 000的Western blotting鉴定结果Fig.2 Western blotting of serum titered at 1∶32 000
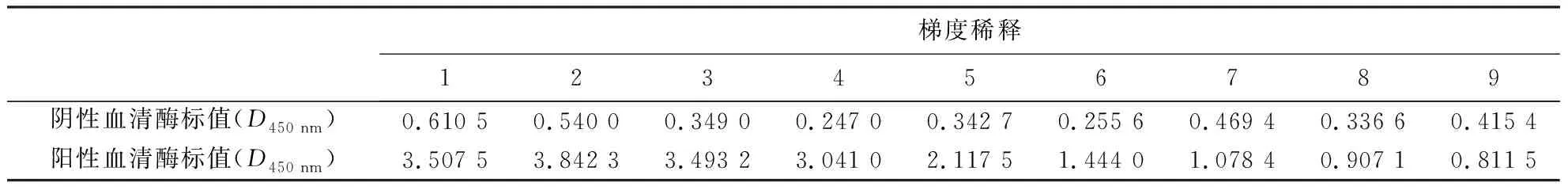
表1 阳性血清效价的ELISA检测结果1)Table 1 ELISA test of positive serum
2.3 杂交瘤细胞筛选结果与融合细胞上清效价检测结果
对融合细胞上清进行ELISA检测,以待检孔值∶阴性孔值≥2判定为阳性,挑选比值高的阳性孔进行3次克隆化.第1次克隆化检测到12孔的阳性,结果如表2所示;第2次克隆化检测到6孔的阳性,结果如表3所示;第3次克隆化检测到2孔的阳性,结果如表4所示.3次克隆化检测结果表明,总共筛选到2孔阳性,分别是3-3B和3-7B.

表3 第2次克隆化检测的阳性结果Table 3 Positive selection after 2nd cloning

表4 第3次克隆化检测的阳性结果Table 4 Positive selection after 3rd cloning
2.4 mAb与p10.8蛋白的特异性鉴定结果
将第3次克隆化检测为阳性孔的3-3B和3-7B上清做1∶2 000稀释,然后均与原核表达和真核表达的p10.8蛋白进行Western blotting检测,同时设置His和Flag标签蛋白作为对照.结果显示,只有3-7B孔的上清能同p10.8蛋白发生特异性反应,将此杂交瘤细胞株命名为H3-7B株.图3显示,H3-7B株mAb能与p10.8原核表达、真核表达蛋白发生反应.其中,原核表达p10.8蛋白与mAb发生反应后,其特异性条带位于28.8 ku处,与预期结果相符(图3A);与真核表达蛋白发生反应后,特异性条带位于预期的12.8 ku处(图3B).表明该H3-7B株mAb是p10.8的mAb.

A:细胞上清与原核表达蛋白的Western blotting鉴定(1:PBS空白对照组;2:p10.8原核表达蛋白组);B:细胞上清与真核表达蛋白的Western blotting鉴定(1:空细胞对照组;2:p10.8真核表达蛋白组).图3 mAb与p10.8蛋白的特异性鉴定结果Fig.3 Specificity identification of mAb and p10.8 protein
2.5 mAb与MDRV、禽腺病毒、鸭坦步苏病毒全病毒的反应结果
图4显示,杂交瘤细胞H3-7B株上清能与MDRV全病毒发生反应,而不能与禽腺病毒、鸭坦步苏病毒全病毒发生反应,表明该mAb对MDRV病毒具有良好的特异性.

图4 3种病毒与杂交瘤细胞上清的反应性鉴定结果Fig.4 Reactivity identification of 3 viruses and hybridoma supernatants
2.6 mAb效价及亚类鉴定结果
对mAb的亚类进行分析,测定酶标值(D450 nm).结果(表5)表明,H3-7B株mAb亚型为IgG2a类.

表5 mAb亚类鉴定结果1)Table 5 Identification of mAb subclass
3 讨论
MDRV感染后侵袭的器官主要是肝脏和脾脏,以靶器官出现灰黄、灰白色坏死点为主要临床特征[19].MDRV有多个开放阅读框(ORF),编码多种蛋白,共同参与病毒的复制和感染,其中,p10.8蛋白是导致该病的重要蛋白之一.p10.8蛋白也是一种可诱导细胞凋亡的非结构蛋白,由MDRV S4基因片段的ORF1编码[20-21].研究表明,p10.8蛋白能够通过内质网应激等多种途径诱导细胞发生凋亡和周期停滞[22],然而目前仍然多采用真核表达载体上的标签蛋白抗体来进行研究.因此,制备抗MDRV p10.8的抗体对于MDRV分子致病机制的研究具有十分重要的意义.
本试验采用常规免疫法对小鼠进行3次免疫,第4次免疫后取阳性血清效价高的脾细胞与SP2/0进行融合,并用间接ELISA法检测细胞上清的效价,连续亚克隆,获得高效价的mAb H3-7B株,该抗体特异性强.陈桂华等[23]在制备猪圆环病毒3型(PCV3)Cap蛋白的mAb时,将抗PCV3 Cap蛋白mAb和抗His标签抗体分别作为一抗进行Western blotting鉴定,并在相应位置出现特异性条带.唐泽群等[24]采用chTLR3重组真核质粒为免疫原,制备了抗chTLR3的mAb,并用原核蛋白等进行生物学活性特异性鉴定.贾晓雪等[25]在制备和鉴定抗牛传染性鼻气管炎病毒(IBRV)gD蛋白mAb时,曾用IBRV全病毒和重组蛋白对抗体特异性进行鉴定.本试验则采用重组原核表达蛋白作为免疫原制备mAb,用Western blotting法对重组原核蛋白、重组真核蛋白与两者相应的标签蛋白His、Flag进行特异性鉴定,并对MDRV全病毒均进行了特异性鉴定,mAb均发生了良好的特异性反应,也证明了本试验所制备的mAb具有良好的生物学活性,且用重组真核蛋白和全病毒抗原检测可以更好地排除His标签所带来的干扰.可见,本试验制备的mAb具有良好的特异性.
