携带FAT1-FAT2基因打靶载体构建及其在猪肾细胞的表达与功能鉴定
2021-10-05李紫聪
林 纯,汤 飞,李紫聪
(1.广东岭南职业技术学院护理与健康学院,广东 广州 510663;2.中山大学华南生物安全四级实验室(筹),广东 广州 510275;3.华南农业大学动物科学学院/国家生猪种业工程技术研究中心,广东 广州 510642)
【研究意义】ω-3多不饱和脂肪酸(PUFAs)是一类具有广泛生物活性的物质,对于防治人类心血管疾病、癌症、糖尿病等具有积极作用,在临床上已被证明具有较好的治疗效果[1-3]。由于猪体内缺乏合成两种主要PUFAs(α-亚麻酸和亚油酸)所必需的ω-3多不饱和脂肪酸脱氢酶基因和Δ-12脂肪酸去饱和酶基因[4-5],导致整个PUFAs的合成途径受到极大限制。通过基因工程和转基因技术来改变猪体内自身合成多不饱和脂肪酸的途径[6],有望改变和提高猪肉中多不饱和脂肪酸的种类及含量,则人类在食用猪肉的同时还能补充必需的多不饱和脂肪酸,对人类的营养和健康具有积极意义,而且还可以预防心血管、关节炎等疾病发生[7]。
【前人研究进展】FAT1基因来源于秀丽隐杆线虫,翻译产物是ω-3多不饱和脂肪酸脱氢酶,该基因是合成ω-3 PUFAs的关键[8]。2006年Lai等在美国报道了首例转线虫FAT1基因的克隆猪诞生[9-10];研究发现,转FAT1基因猪的总ω-3 PUFAs含量比对照高了3倍多,ω-6 PUFAs含量下降了23%,并且ω-6/ω-3比例相对于对照从8.52下降到1.69;肌肉、肝脏、肾脏等主要组织中的ω-3 PUFAs含量均有所提高,揭示了FAT1转基因猪能产生富含ω-3 PUFAs的猪肉,可大大提高猪肉的风味及营养保健价值[7]。FAT2基因同样来源于线虫,是编码Δ-12脂肪酸去饱和酶的关键基因,是合成ω-6 PUFAs的关键。2008年中国农业大学孔平等[11]报道成功克隆了FAT2基因,并利用哺乳动物的密码子偏好性进行优化,构建真核表达载体,通过细胞稳定转染筛选获得阳性细胞群;经过在细胞水平上的分子生物学测定分析表明,FAT2基因稳定整合到CHO细胞基因组并表达,使哺乳动物细胞中的亚油酸含量显著提高[12]。目前,去饱和酶基因的克隆主要以真菌、酵母、藻类、植物、昆虫等为资源,其中可从藻类中克隆Δ-12、Δ-9、Δ-6、ω-3基因;从拟南芥、菠菜中克隆FAD2、FAD3基因[13];从大豆中克隆Gm FAD3基因[14]。这些基因很多已被广泛应用于转基因试验,以此来改良作物的油脂组成,但应用于转基因动物特别是转基因猪上的研究相对比较少。
【本研究切入点】关于ω-3多不饱和脂肪酸脱氢酶和Δ-12脂肪酸去饱和酶,前人大多数克隆单个基因进行表达,且只致力于重建ω-6PUFAs转变成ω-3 PUFAs的通路[15-17]。哺乳动物体内缺乏Δ-12脂肪酸去饱和酶,因此ω-6PUFAs含量稀少。本研究拟克隆编码FAT1和FAT2基因,构建油酸转变为ω-6PUFAs以及ω-6PUFAs转变成ω-3 PUFAs的两个通路,通过转基因技术将FAT1、FAT2基因稳定转入动物体内进行表达以达到更好效果。【拟解决的关键问题】克隆获得FAT1和FAT2基因,经密码子优化后构建携带FAT1-FAT2双基因的打靶载体[18],载体含以rRNA基因间内部转录间隔序列为靶位点序列的DS1、DS2,NEO与EGFP融合的正筛选系统[19-20],TK基因的负筛选系统,以及Cre-loxp删除系统。将构建好的打靶载体转染猪肾细胞PK15,验证目的基因在细胞水平的表达及检测细胞中脂肪酸的相对含量。
1 材料与方法
1.1 试验材料
E.coliDH5a感受态细胞(TaKaRa)购自广州瑞真生物技术有限公司;载体pOSDupDel购自华灿生物科技有限公司;质粒PF1、PUC57-2AFAT2、PCMMPD-DS1/2、PN1由华南农业大学动物科学学院李紫聪教授课题组构建并保存。猪肾细胞PK15采自广东温氏食品集团华农温氏股份有限公司水台原种猪场。
主要试剂:TaKaRa LA Taq with GC buffer、PrimeSTAR HS DNA Polymerase、Ligation Kit Ver.2.0、In-Fusion® Advantage PCR Cloning Kit(Clontech)、反转录试剂盒PrimeScript® RT reagent Kit with gDNA Eraser(TaKaRa)均购自广州瑞真生物技术有限公司,Taq Mix Kit 购自广州东盛生物科技有限公司;限制性内切酶SspⅠ、DrɑⅢ、AscⅠ、PɑsⅠ、PmeⅠ、NruⅠ等购自广州昂科生物技术有限公司;转染试剂盒LipofectamineTM LTX with PLUSTM Reagent(Invitrogen)、G418sulfate(Gibco)、DMEM(Gibco)、FBS(Gibco)、0.25%胰酶(Gibco)、DPBS(Gibco)等均购自广州佰默生物科技有限公司。
主要仪器设备:凝胶成像系统(广州三元科技有限公司)、LongGene PCR仪(M966Y,广州深华科技有限公司);倒置显微镜(TE300、TE2000U,日本Nikon);正置荧光显微镜(E800,日本Nikon)、气相色谱-质谱联用仪(TRACE GC-2000 GC-MSTM,美国Thermo Finnigan)。
1.2 试验方法
1.2.1 目的基因FAT1-2A-FAT2的获取 为了构建脂肪酸去饱和酶双基因FAT1、FAT2联合表达载体,设计一段2A连接肽,先将2A与FAT2进行基因融合,再通过重叠延伸PCR方法将FAT1基因与已融合2A片段的FAT2基因连接起来,在重叠基因两端加上酶切位点PɑsⅠ和PmeⅠ,将双基因同时连接到打靶载体上。
(1)2A连接肽与FAT2基因的克隆:FAT2基因来源于秀丽隐杆线虫,该基因在NCBI上的登录号为NM-070159,编码区全长1 131 bp,编码含376个氨基酸的蛋白。为使该基因在猪体上高效表达,对所获取的序列根据猪的密码子偏好性进行改造合成。以质粒PUC57-2A-FAT2为模板,设计引物2A-FAT2-F、2A-FAT2-R(表1)扩增得到2A-FAT2片段。对PCR产物纯化回收,待用。
(2)FAT1基因的克隆:以质粒PF1为模板,设计与2A-FAT2片段重叠20 bp左右的PCR引物FAT1-F、FAT1-R(表1)扩增得到FAT1片段。对PCR产物纯化回收,待用。
(3)FAT1与FAT2的重叠延 伸PCR:以FAT1基因与2A-FAT2基因互为共同模板,按照图1重叠PCR设计原理进行扩增。经普通PCR程序进行扩增后纯化回收,待用。
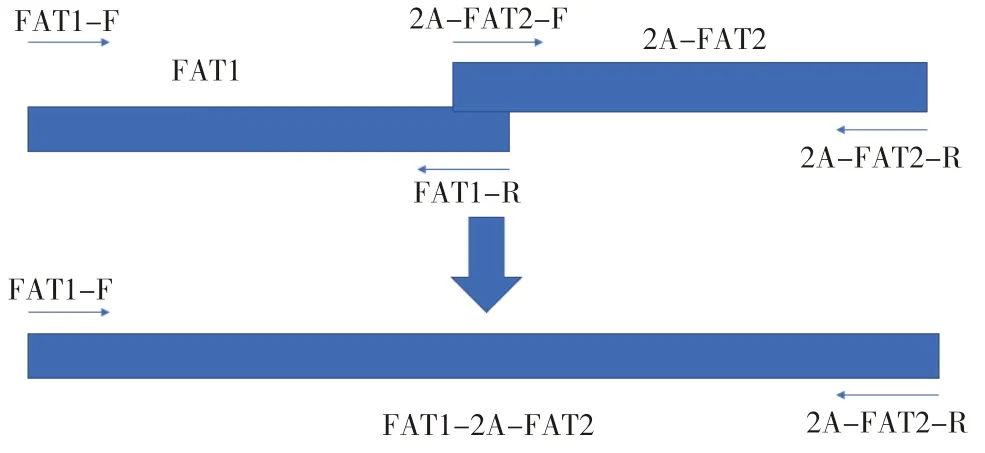
图1 FAT1-FAT2重叠延伸PCR引物设计原理Fig.1 Design principle for PCR primer of FAT1-FAT2 overlapping and extended
1.2.2 质粒P-D1-FAT1FAT2-D2的构建 设计与PF1载体酶切位点PɑsⅠ和PmeⅠ两端重叠15 bp的引物F、R(表1),以纯化回收的FAT1-2AFAT2基因为模板,进行PCR扩增FAT1-2A-FAT2基因。配制酶切体系,利用PɑsⅠ和PmeⅠ对载体PF1进行双酶切,再把重叠基因FAT1-2AFAT2与打靶载体进行连接,构建质粒P-D1-FAT1FAT2-D2。将连接产物转化大肠杆菌,进行菌液PCR,将验证正确的菌液送至上海英骏生物技术有限公司进行测序,验证插入目的基因的正确性。将测序正确的菌液扩繁后进行质粒抽提,酶切鉴定后保种,待用。

表1 PCR引物信息Table 1 Information of PCR primers
1.2.3 重组质粒PF1F2的构建 本研究的打靶载体属于置换型,载体上的同源重组引导序列与基因组正常方向一致,通过双交换使外源DNA替换相对应的基因组内同源序列,由此将线性化的载体置换到基因组内。为了使打靶载体发挥更好的作用及便于后期对中靶细胞进行检测,一般打靶载体使用长短臂[2-3]。将已构建好的质粒P-D1-FAT1FAT2-D2中长约1.1 kb的HRDS1臂置换为长为2.0 kb的DS1(L)。为方便后期在Cre-loxp系统介导下删除抗性基因Amp,在DS1(L)左侧添加1个loxp位点。载体中只有NEO正筛选选择系统,为了富集同源重组的克隆,提高定点整合的效率,添加负筛选基因TK,只有通过正负系统双重选择才能最大程度密集定点整合细胞。重组质粒PF1F2的构建路线如图2所示。
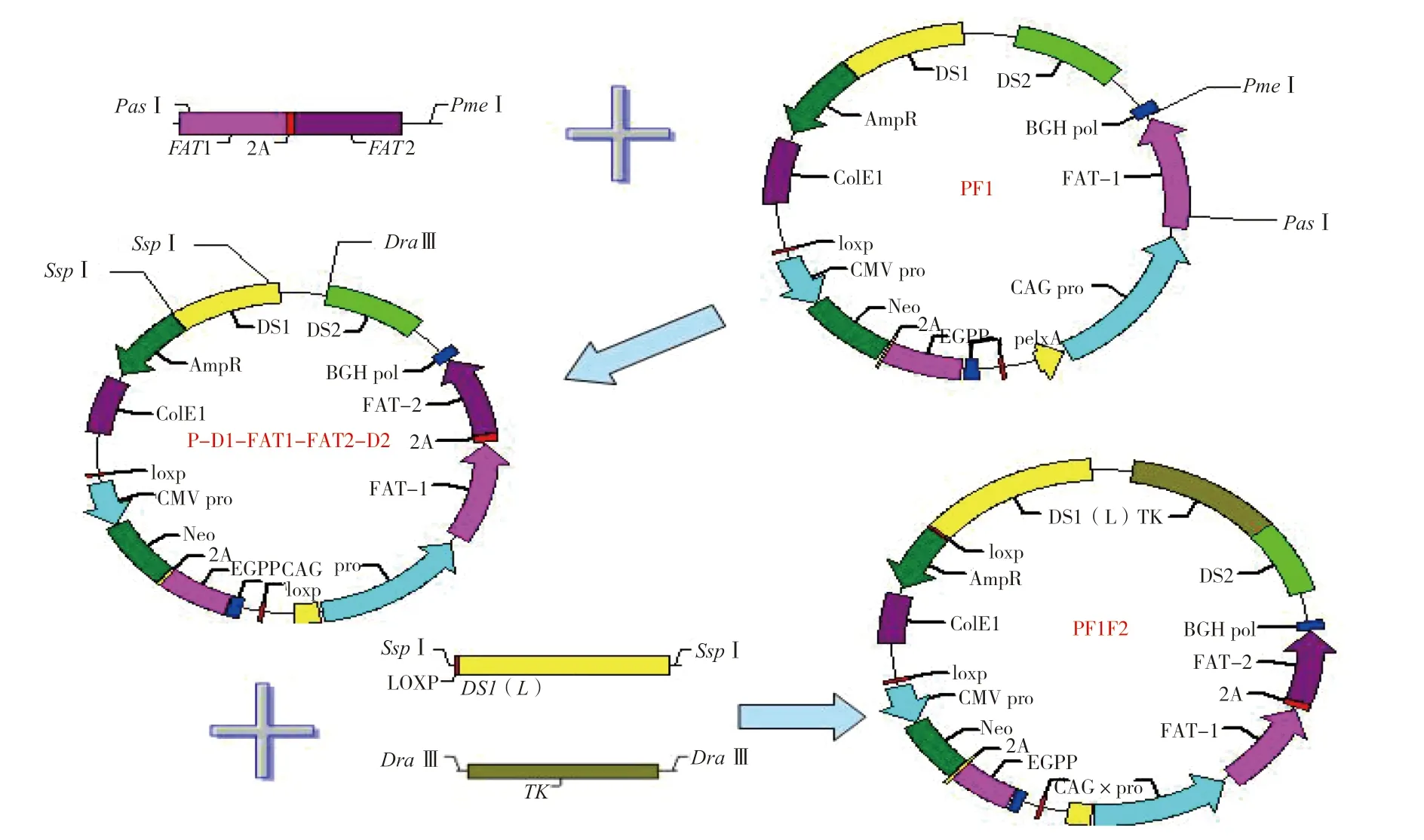
图2 重组质粒PF1F2的构建路线Fig.2 Construction route of recombinant plasmid PF1F2
(1)DS1(L)基因扩增:以质粒PCMMPDGFP-NEO-DS1(L)为模板,设计引物Loxp-DS1(L)-F、Loxp-DS1(L)-F(表1)扩增DS1(L)基因,并在上、下游引物两端加入限制性内切酶SspⅠ,PCR扩增体系及程序同常规。PCR产物检测正确后纯化回收。
(2)TK基因扩增:以载体pOSDupDel为模板,设计引物TK-F、TK-R(表1)扩增TK基因,并在上、下游引物两端加入限制性内切酶DrɑⅢ,PCR扩增体系及程序同常规。PCR产物检测正确后纯化回收。
(3)DS1(L)、TK基因与载 体P-D1-FAT1FAT2-D2连接:参考构建上述质粒的方法,通过设计DrɑⅢ、SspⅠ酶切位点,回收纯化DS1(L)基因、TK基因的PCR产物,与P-D1-FAT1FAT2-D2进行连接、转化。挑取转化后的白色单菌落进行培养,以菌液为模板,进行菌液PCR检测,测序鉴定等步骤,构建得到重组质粒PF1F2。抽提质粒,酶切鉴定后保种,待转染细胞。为方便对TK基因进行PCR鉴定,设计引物TK(825)-F、TK(825)-R,(表1),扩增部分TK基因(长度825 bp)。
1.2.4 细胞转染 对液氮中取出的PK15细胞系复苏,细胞培养液为含10%胎牛血清的高糖DMEM。介导转染前将细胞传至六孔板中,待细胞长至50%~80%时,将经过去内毒素提取的对照质粒PN1、PF1、PF1F2分别利用脂质体介导的方法转染PK15细胞,转染后在荧光显微镜下观察荧光表达情况。
1.2.5RT-PCR检测基因表达 分别收集质粒转染后的细胞提取RNA,进行RT-PCR检测。FAT1-FAT2双基因PCR检测引物序列为F:5'GGAGAAGGAGGCCCCCAGGGACGT3',R:5'CTGATCAGCGGGTTTAAACAAAG3';设计跨过内含子的β-ɑctin引物,作为阳性对照,引物序列为β-ɑctin-F:5'ACCCCAAAGCCAACCGTGAG 3',β-ɑctin-R:5'AAGCGCTCGTTGCCGATGG 3'。
1.2.6 脂肪酸分析 分别收集质粒转染后的细胞,转移到离心管中,用DPBS缓冲液洗涤细胞后离心,吸除多余水分,加入1 mL 2.5% H2SO4甲醇溶液进行甲酯化处理,充分裂解细胞,80 ℃加热90 min;待冷却至室温,加入1.5 mL 0.9%NaCl溶液和1 mL正乙烷,震动、低速离心(2 000~3 000 r/min);离心后取含有甲酯的上层溶液用于GC-MS气质联用分析检测。
GC-MS气质联用分析条件:色谱柱DB-5,30 m×0.25 mm;恒流进样,进样体积1 μL;进样口温度220 ℃;升温程序为100 ℃ 5 min(10 ℃/min)→200 ℃ 5min(5 ℃/min)→220 ℃10 min;载气He,流速1.0 mL/min;EI+离子源,质谱轰击电子能量为70 eV、350V;质量扫描范围35~325 amu。谱库检索:MassLynx和NIST。环境条件:室温20 ℃,相对湿度35%。
2 结果与分析
2.1 目的基因FAT1-2A-FAT2的获取
通过PCR分别扩增FAT1、2A-FAT2基因,产物用1%琼脂糖凝胶进行电泳,可见到特异性扩增条带,产物长度分别为1 238、1 218 bp(图3),大小与预期相符。以FAT1基因与2A-FAT2基因互为共同模板,进行FAT1与FAT2的重叠延伸PCR,重叠延伸PCR产物用1%琼脂糖凝胶进行电泳检测,得到2 426 bp条带(图3),与预期的重叠片段相符,证明成功延伸出重叠片段。
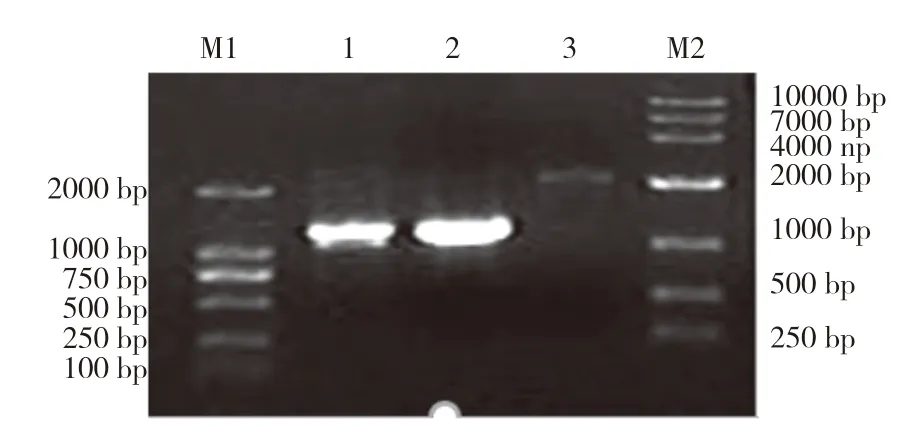
图3 FAT1、2A-FAT2、FAT1-2A-FAT2基因扩增结果Fig.3 Amplification results of FAT1,2A-FAT2 and FAT1-2A-FAT2 genes
2.2 质粒P-D1-FAT1FAT2-D2的构建
利用与载体PF1酶切位点PɑsⅠ和PmeⅠ两端重叠15 bp的引物,以纯化回收的FAT1-2AFAT2基因为模板,扩增重叠基因并纯化回收后,利用限制性酶切位点PɑsⅠ和PmeⅠ对打靶载体PF1进行双酶切,经连接、转化,构建成P-D1-FAT1FAT2-D2质粒。
质粒P-D1-FAT1-FAT2-D2经PCR鉴定显示,扩增得到大小为2 439 bp的片段,与预期相符(图4);用PmeⅠ和BstBⅠ酶切消化质粒P-D1-FAT1-FAT2-D2,BstBⅠ为FAT1与FAT2基因中间的1个单一限制性酶切位点,酶切后质粒产生与预期相符的两条带,一条为插入的目的序列,大小约为1 208 bp;另一条为载体序列,大小约为11 339 bp(图5)。而测序结果比对同源性100%,证明重叠基因FAT1-2A-FAT2已经连接到载体上,而且片段编码区无突变。
图4 质粒P-D1-FAT1FAT2-D2 PCR鉴定Fig.4 PCR identification of plasmid P-D1-FAT1FAT2-D2
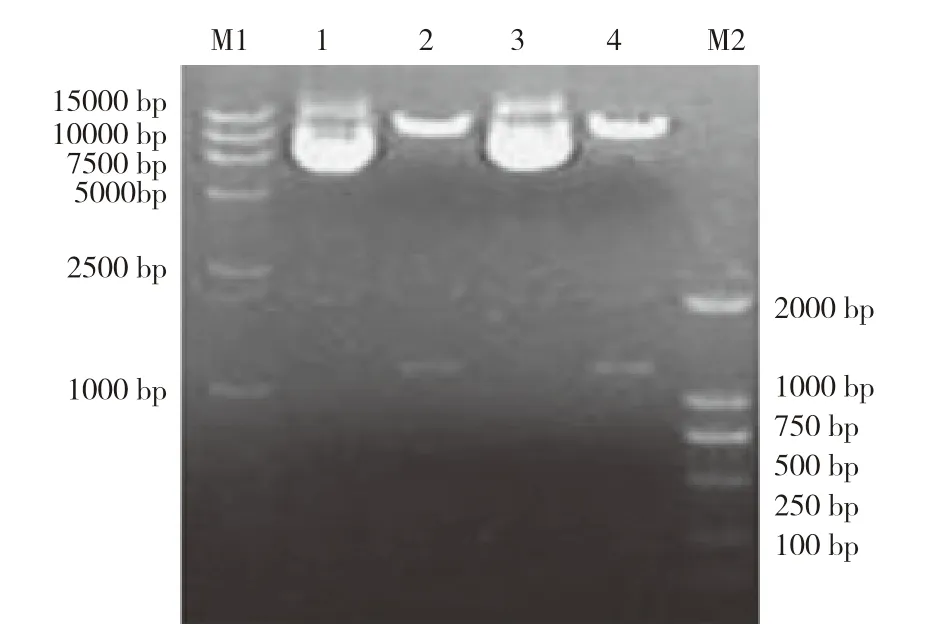
图5 质粒P-D1-FAT1FAT2-D2酶切鉴定Fig.5 Digestion identification of plasmid P-D1-FAT1FAT2-D2
2.3 重组质粒PF1F2的构建
通过PCR分别扩增DS1(L)、TK基因,产物用1%琼脂糖凝胶进行电泳,可见到特异性扩增条带。DS1(L)基因扩增产物长度为2 030 bp,TK基因扩增产物长度为1 730 bp(图6),大小与预期结果相符。
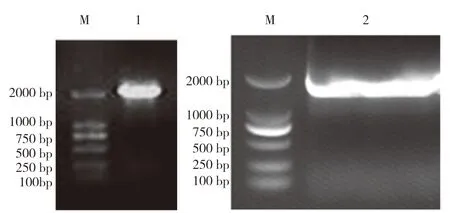
图6 DS1(L)、TK基因扩增结果Fig.6 Amplification results of DS1(L)and TK genes
将DS1(L)、TK基 因PCR产物与载 体P-D1-FAT1FAT2-D2进行连接、转化。用DrɑⅢ酶切质粒PF1F2,酶切后质粒产生与预期相符的两条带,一条为插入的TK序列,大小为1 730 bp;另一条为载体片段,大小约为13 477 bp(图7)。将上述鉴定正确的菌液送到上海英骏生物技术有限公司进行测序,测序结果比对同源性为100%,证明DS1(L)、TK基因均已经连接到载体上,而且片段编码区无突变。
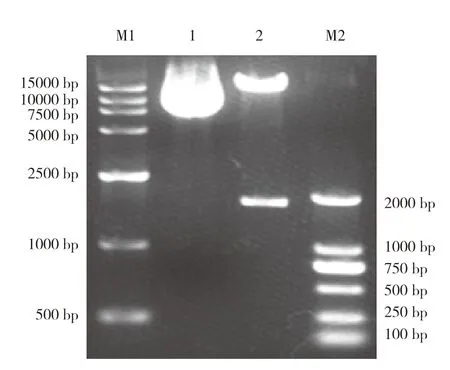
图7 PF1F2质粒酶切鉴定Fig.7 Digestion identification of PF1F2 plasmid
2.4 细胞转染
重组质粒PF1F2转染PK15细胞48 h后,在荧光显微镜下可看到转染阳性率约为50%的荧光表达细胞(图8)。

图8 质粒PF1F2转染48 h后的PK15细胞Fig.8 PK15 cells transfected with PF1F2 for 48 h
2.5 RT-PCR检测基因表达
收集质粒转染后的PK15细胞,用试剂盒提取细胞总RNA,以总RNA为模板进行反转录,以目的基因FAT1-FAT2的特异性引物进行PCR,同时以β-ɑctin基因作为阳性对照,用于β-ɑctin基因PCR扩增的引物跨过内含子,引物扩增β-ɑctincDNA的片段大小为434 bp,目的基因扩增的片段大小为2 439 bp。由图9可见,反转录产物中无DNA污染,目的基因在细胞中表达,阴性对照无检测到基因表达。

图9 RT-PCR检测基因表达结果Fig.9 Result of gene expression detected by RT-PCR
2.6 脂肪酸测定结果
瞬时转染细胞48 h后收集细胞,提取总脂肪酸进行GC-MS气质联用分析脂肪酸含量[17]。利用GC-MS法对对照(转染PN1空载体)和两个试验组(转染PF1,转染PF1F2质粒)的脂肪酸含量进行比较。
由表2可知,转染后,与对照相比,属于ω-6 PUFAs的亚油酸(C18:2n-6)、花生四烯酸(C20:4n-6)以及总体的ω-6PUFAs含量在试验组中均下降,特别在转染FAT1-FAT2双基因的试验组中下降比例更加显著;属于ω-3 PUFAs的亚麻酸(C18:3n-3)、EPA(C20:5n-3)、DHA(C22:6n-3)以及总体的ω-3PUFAs含量在试验组中呈上升趋势,且在转染PF1F2质粒的试验组中3种不饱和脂肪酸含量相比对照存在显著差异。ω-6/ω-3的比例由对照的1.4显著下降到试验组的0.42~1。ω-6PUFAs与ω-3PUFAs之间此消彼长的关系正是因为FAT1、FAT2基因表达起作用的结果。当两个基因同时发挥作用时,生成的ω-3 PUFAs含量更多,效果更加明显。
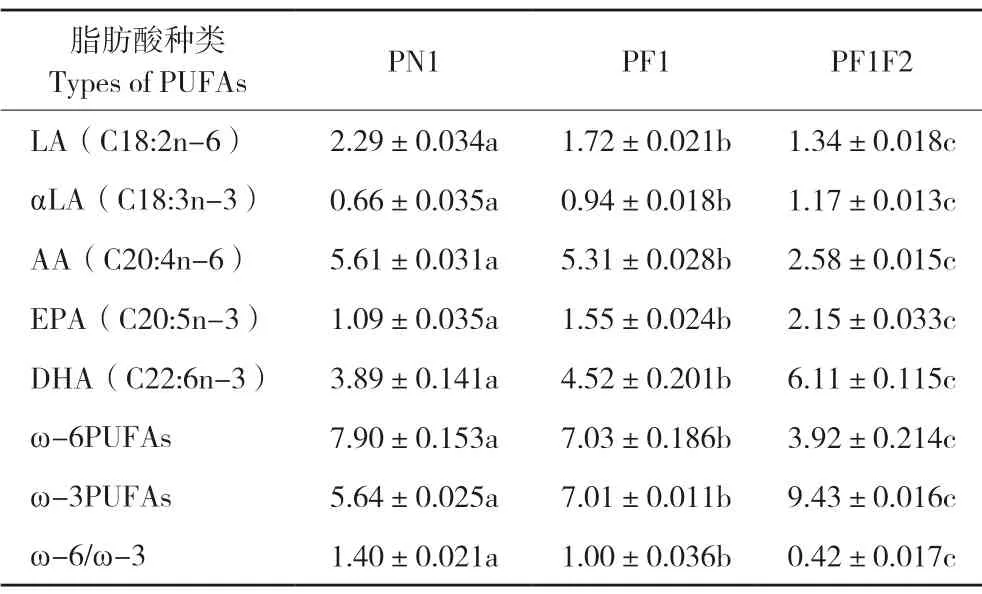
表2 瞬时转染PK细胞的脂肪酸组成Table 2 Fatty acid composition of transient transfected PK15 cells(%)
3 讨论
由于哺乳动物体内缺乏ω-3多不饱和脂肪酸脱氢酶关键基因FAT1、Δ-12脂肪酸去饱和酶的关键基因FAT2,因此不能自身合成亚麻酸、亚油酸等。本研究通过构建携带FAT1-FAT2双基因的打靶载体,通过基因表达,FAT1基因能够发挥其ω-3不饱和脂肪酸脱氢酶的作用,将ω-6系列不饱和脂肪酸转变为相应的ω-3不饱和脂肪酸;FAT2基因也能够发挥其Δ-12去饱和酶的作用,将动物体内含量较多的油酸转变为ω-6不饱和脂肪酸。在这两个基因的协同作用下,最终使得ω-6 PUFAs含量降低,而ω-3 PUFAs含量提高。
本试验中,ω-3 PUFAs含量提高的幅度相对较低,这与转染效率不高有一定关系,而且利用细胞测定脂肪酸含量过程中,部分脂肪酸由于样本含量小、信号弱、不灵敏而无法检测,因此未能显示出所有脂肪酸组分,导致测定结果存在一定偏差。此外,细胞培养液成分含有亚油酸,如胎牛血清中含有少量亚油酸,马血清中含有大量亚油酸,一定程度上会改变细胞中的脂肪酸组成。因此为了保证试验结果的准确性,本试验过程中使用同一批次的胎牛血清,并且严格按照10%的浓度添加到培养基中。本试验还利用了以猪rRNA基因间的内部转录间隔序列ITS为靶位点,使基因打靶的靶位点增加到100~300个,从而使基因定点整合效率大幅提高。后续研究可筛选获得定点整合FAT1-FAT2基因的转基因细胞系,通过核移植生产转FAT1-FAT2基因克隆猪,在猪体内重建生产多不饱和脂肪酸的途径,从而提高猪肉的营养和保健价值。
4 结论
本研究成功构建了含NEO与EGFP融合基因及TK基因的正负筛选系统,以及Cre-loxp删除系统的FAT1-FAT2双基因多位点打靶载体PF1F2。载体PF1F2转染细胞后,FAT1、FAT2基因可以正常表达并分别发挥ω-3多不饱和脂肪酸脱氢酶和Δ-12脂肪酸去饱和酶的作用,显著降低细胞中ω-6 PUFAs含量,提高ω-3 PUFAs含量,降低ω-6/ω-3的比例。
