提升燃料电池铂基催化剂稳定性的原理、策略与方法
2021-09-28梁嘉顺刘轩李箐
梁嘉顺,刘轩,李箐
华中科技大学材料科学与工程学院,材料成型与模具技术国家重点实验室,武汉 430074
1 引言
质子交换膜燃料电池(Proton exchange membrane fuel cell,PEMFC)是一种高效、清洁的能源转换装置,可以通过电化学的方式将储存在氢气和氧气中的化学能转化为电能1-4。PEMFC具有十分广泛的应用前景,包括基站式电源、便携式电源以及电动汽车等。例如,2015年丰田公司推出了量产的氢燃料电池轿车—Mirai,极大地推动了PEMFC的应用5-7。然而目前PEMFC的大规模使用仍然受到其高成本、低使用寿命等因素的限制。PEMFC阴极发生的氧气还原反应(Oxygen reduction reaction,ORR)在动力学上十分缓慢,需要大量的贵金属铂(Pt)作为催化剂来加速反应的进行8-10。例如上述提到的Mirai汽车阴极使用的碳(C)载PtCo/C催化剂,阴极和阳极(催化剂为Pt/C)的Pt负载量分别为0.315和0.05 mg·cm-2,远高于美国能源部2020年的技术指标(0.1和0.025 mg cm-2)11,12。
在过去的十年内,Pt基氧还原催化剂的研究取得了较大的进展,研究者们通过密度泛函理论(DFT)计算搭建了催化剂电子结构-ORR活性的理论模型,即适当降低催化剂表面Pt原子的d-带重心能够削弱铂与含氧中间体的结合能,从而加快反应中间体的脱附来提高催化剂的活性13-16。基于这一理论模型,研究者在实验上设计了许多的铂基合金催化剂,包括Pt-Ni、Pt-Co以及Pt-Fe等来提高催化剂的氧还原活性17-19,并且取得较好的结果。然而这些合金催化剂通常存在稳定性不足的问题,这是因为在质子交换膜燃料电池高电压(0.6-1.0 V)和酸性环境的工作条件下,非贵金属极易发生溶解流失,导致催化剂性能的下降。燃料电池要想大规模的应用,必须具备良好的运行稳定性,我国科技部以及美国能源部针对燃料电池/氧还原催化剂稳定性提出的技术指标为:0.6-0.95 V电势循环30000圈后,活性衰减小于40%以及1.0-1.5 V电势循环5000圈后,活性衰减小于40%。
在本文中,我们将聚焦于PEMFC/氧还原催化剂稳定性的问题,总结目前提升催化剂稳定性的一些原理、策略与方法。首先我们将从热力学、动力学原理上阐述影响催化剂稳定性的原因及其调控原理。随后,我们将概述一些具有代表性的提升催化剂稳定性的策略和方法。最后,我们将讨论目前仍存在的一些问题以及将来可能的研究方向。
2 提升稳定性的热力学、动力学原理
ORR涉及到多个质子/电子的转移,反应过程较为复杂。通过两电子转移,氧气可以部分还原得到过氧化氢;而通过四电子转移过程,氧气则可以完全反应得到水,其反应路径如下所示:
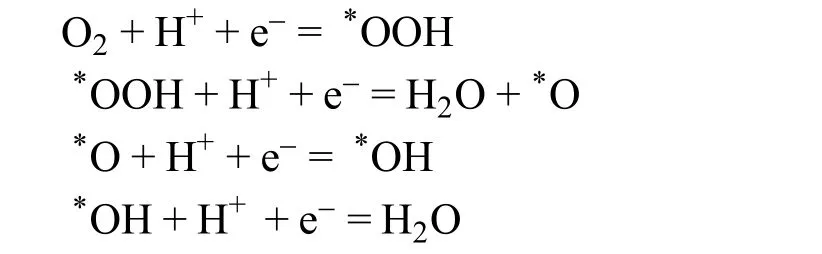
对于这些多电子转移步骤,中间会存在较多的反应中间体,例如O*、HO*、和HOO*等(其中*代表吸附在催化剂表面的物种)。Sabatier原理指出,一个优秀的催化剂需要对反应物种具有适中的吸附能,从而平衡反应物种在催化剂上的吸附和脱附过程20,21。理论研究表明,Pt与含氧中间体的结合能较强,需要对其电子结构进行调控来提高催化剂的ORR活性13。
为了提高Pt催化剂的ORR活性,研究者一般会将Pt和其他过渡金属(M),例如Fe、Co、Ni等后期过渡金属进行合金化。这是因为,一方面由于M与Pt具有不同的电子结构(例如费米能级不同,投影态密度不同等),Pt与邻近原子之间存在电荷转移的效应,可以称之为配体效应(Ligand effect)13,16;另一方面,M的原子直径一般比Pt原子小,因此合金化会导致晶格收缩,Pt原子层会受到压缩应力,使Pt的d带变宽,即应力效应22,23。这两种效应都能够使Pt原子的d带重心下移,弱化Pt与含氧中间体的结合能,提高催化活性。因此,目前比较常见的高活性催化剂均为Pt基合金催化剂。
然而,燃料电池的工作条件为高电压(0.6-1.0 V)以及强酸性(pH < 1),在这种工作条件下,催化剂中的过渡金属极易发生氧化、溶解流失,导致合金化带来的配体效应、应力效应的消失,催化活性下降;而且,溶解的过渡金属离子容易与质子交换膜发生反应,导致其电导率降低甚至发生降解;另一方面,Pt原子的溶解流失,以及溶解再沉积(电化学熟化)也将导致催化剂活性的下降24-26。因此,从这方面看,提高催化剂以及燃料电池的稳定性关键在于,提高催化剂中的Pt以及M的抗氧化腐蚀能力。为了能够更好地设计有效的策略来提高过渡金属的稳定性,我们需要更深入的了解影响金属溶解的热力学和动力学等因素。
2.1 热力学影响因素
从热力学上看,金属的氧化、溶解可以通过其标准电极电势(E0)初步进行判断,当金属所处电势超过其标准电极电势时,金属存在氧化的趋势。表1列出了一些常见金属的标准电极电势,注意这里所列出的标准电极电势为标准状态,即一个大气压下、温度为25 °C且金属离子活度为1时,相对于标准氢电极电势的值。当进一步考虑到pH值对电极电势的影响,我们可以得到pH-电极电势图,又称作布拜图(Pourbaix diagram)27,28。布拜图中明确地示出在某一电位和pH条件下,体系的稳定物态或平衡物态,因此根据布拜图可从热力学上很方便地判断在一定的电位和pH条件下,金属材料发生腐蚀的可能性。例如Pourbaix等27,28报道了铂-水体系的布拜图(图1),从该图中可以看出,Pt在较大电势范围内相对稳定,仅仅在电势为1.0-1.2 V、pH值为-2 - 0的范围内存在少量溶解。
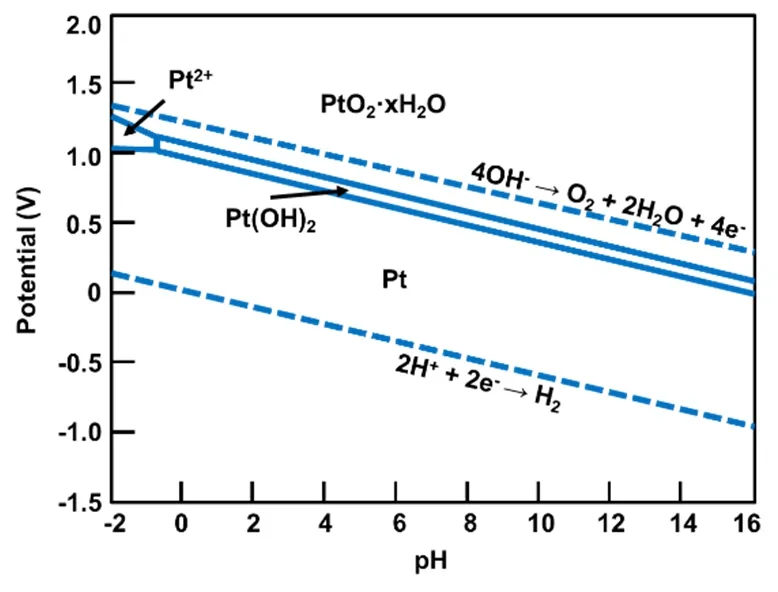
图1 水溶液中Pt物种的布拜图27,28Fig. 1 Pourbaix diagram of Platinum in aqueous solution 27,28.
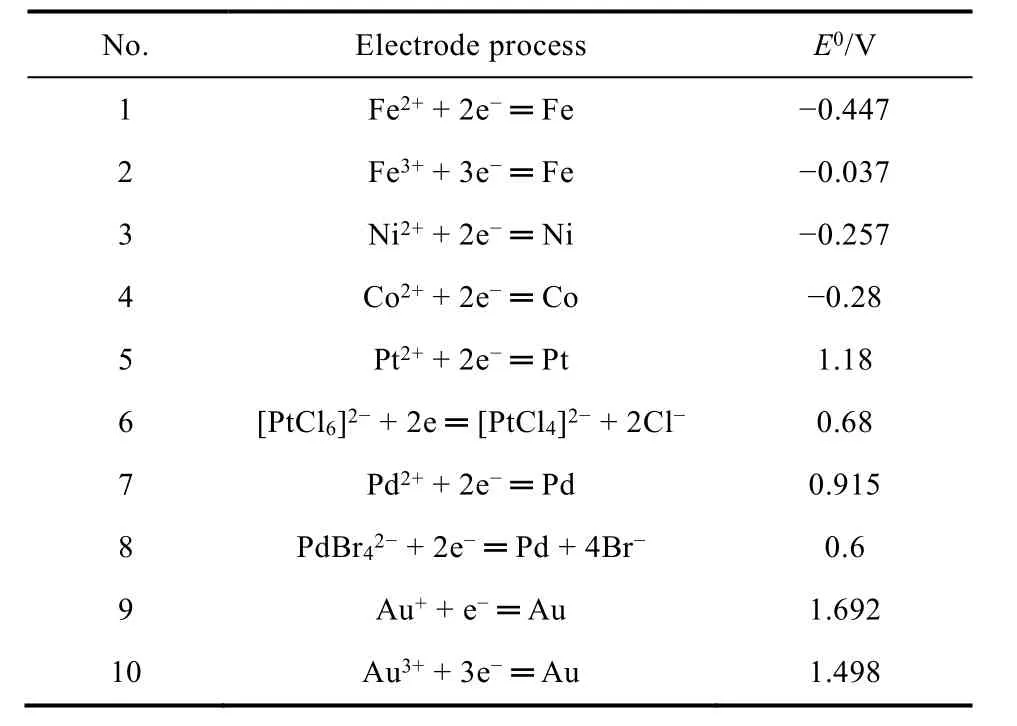
表1 一些常见金属的标准电极电势(25.0 °C,101.325 kPa)Table 1 Standard electrode potential of some metals(25.0 °C, 101.325 kPa).
然而以上的电极电势主要是针对块体材料,而目前常用的催化剂尺寸都在纳米级别,金属的物理化学性质会有较大的变化,因此需要对上述的电极电势做出一些修正。
Jinnouchi等29采用DFT计算了Pt原子溶解电势与一些参数,例如内聚能、配位数、粒子尺寸的关系。根据Gibbs-Thomson方程可以推导出,纳米尺度下Pt原子的溶解电势为:
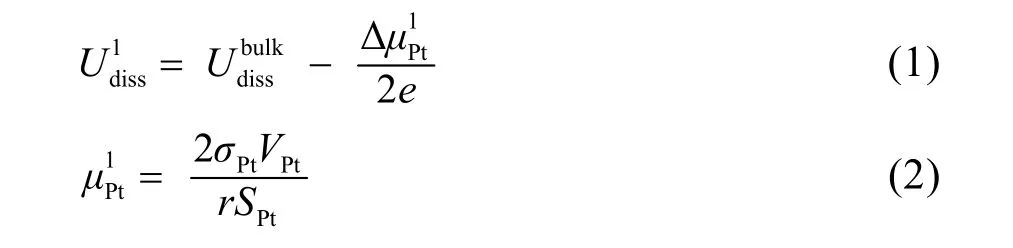
其中,上标1代表单个Pt原子溶解,bulk代表块体材料,σPt、VPt、SPt分别为纳米晶的表面能、单个Pt原子的体积以及表面积,r为纳米晶的半径。另外,作者还进一步考虑了所有Pt原子都溶解的情况,其表达式与上述方程(1)类似,仅在系数上有所不同,因此我们在这里仅以单个原子溶解的情况作进一步讨论。
从上述的方程(1)可以看出,Pt原子的溶解电势与纳米粒子的尺寸、表面能直接相关。当纳米粒子尺寸减小时,其表面能也会增加,Pt原子的溶解电势会急剧下降,导致纳米晶可能在催化过程的溶解、失活。另外,不同的位点、晶面具有不同的表面能,一般来说表面能与配位数成负相关。对于纳米晶边界(edge)、顶点(corner)等低配位的位点在热力学上较不稳定,一般溶解也会从这些位点开始。图2展示了DFT计算得出的纳米晶溶解电势与尺寸、配位数的关系。
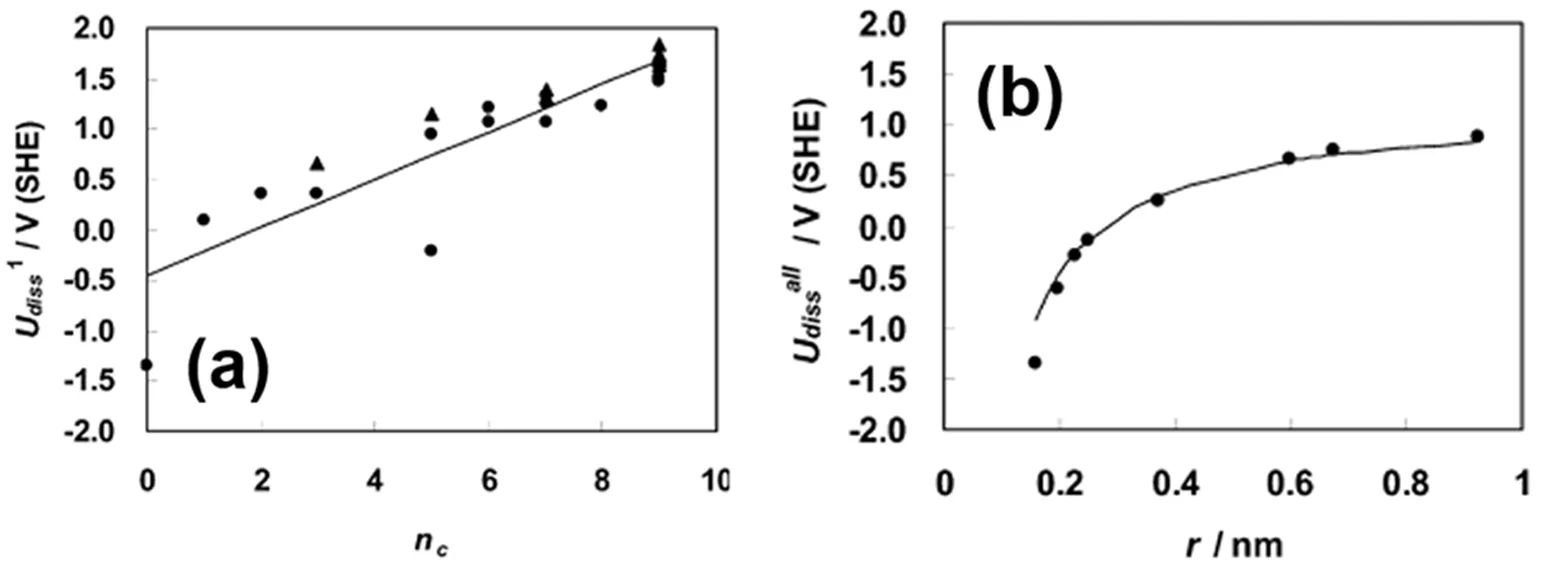
图2 铂原子的溶解电势与配位数(a)、粒子尺寸(b)铂原子的关系29Fig. 2 The relationship between dissolution potential of Pt and coordinate number (a), particle size (b) 29.
从另一个角度看,金属的溶解会受到周围原子的限制,即溶解原子与周围原子的结合能(Eb)、或纳米晶整体的内聚能Ecoh也将对金属溶解电势产生一定影响,其中内聚能(cohesive energy)指凝聚态物质消除分子间作用力,将组成原子拆分成气化单独原子所需要的能量:
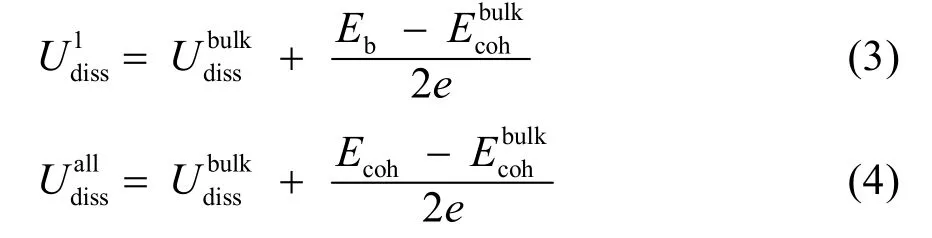
其中上标all代表所有原子均发生溶解的情况。从上述公式(3)和(4)看,提高纳米晶的内聚能或提高目标原子与周围原子的结合能,能够有效提高纳米晶的溶解电势。一般来说,相对于后过渡金属(Fe、Co、Ni等),铂与早期过渡金属(Y、Ce、V等)的合金具有更负的形成焓(标准状态的各种元素的最稳定单质生成标准状态下纯物质的热效应)、以及更高的内聚能,因此,铂-前过渡金属这一系列的催化剂可能具有更好的电催化稳定性30-32。从另一方面看,结合能以及内聚能可以通过键能定性反映,一般Pt―M键与1/2[(Pt―Pt) + (M―M)]的键能差越大,PtM形成焓越负,内聚能越大。因此,可以精细调控纳米晶内Pt―M的键能来提高催化剂的稳定性。
以上的分析主要是基于孤立的纳米粒子,而实际的催化剂是纳米晶分散负载到特定的载体,例如碳粉或其他氧化物上。载体与纳米晶的相互作用,或者称之为金属-载体强相互作用(strong metal-support interactions,SMSI)33,34,也可以影响目标原子与周围原子间的结合能Eb,从而提高纳米晶的稳定性。因此,这也为我们设计催化剂结构,包括界面耦合等,提供一定的理论指导依据。例如,采用氮掺杂碳(N-C)替代传统炭黑作为催化剂载体,通过N-Pt直接的配位作用,能够锚定纳米晶,降低纳米晶团聚、溶解趋势,从而提高催化剂整体稳定性,这一稳定性提升的现象已经在Pt-Co/N-C35,36,Pt-Fe/N-C37等体系中有所报道。
Greeley和Nørskov等38采用DFT计算了多种溶质原子在不同溶剂基底中的溶解电势以及偏析能。如图3所示,溶质的溶解电势似乎存在周期性规律,后期金属溶质(这里指Ni、Cu同族元素)在早期金属溶剂(这里指Fe、Co同族元素)中的溶解电势比标准电极电势高,而早期金属在后期金属中的溶解电势则会降低。有趣的是,他们计算得出的偏析能似乎也存在这样的周期性规律:后期金属溶质在早期金属溶剂中的偏析能更负,更倾向于偏析到表面。实际上,这种关系可以比较好的理解:如果溶质具有很强的偏析趋势,那么该溶质在表面应该在热力学上更稳定,进而具有更强的抗氧化溶解能力。从另一方面看,溶解首先发生在表面,要降低金属的溶解,也可以通过降低金属偏析到表面的趋势来实现。对于Pt基纳米晶催化剂来说,就是要降低M原子的偏析,提高Pt壳层的溶解电势,以此来设计高稳定性的纳米晶催化剂。
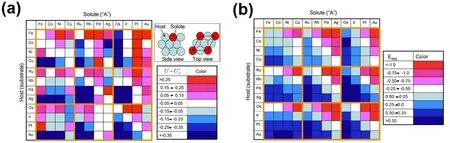
图3 不同溶质在不同溶剂基底中的(a)溶解电势以及(b)偏析能38Fig. 3 Dissolution potential (a) and segregation energy (b) of different solutes in different hosts 38.
2.2 动力学影响因素
对于实际电化学腐蚀过程,需要对体系施加一定的过电位才能驱动反应的进行,这里面就涉及到动力学限制的过程。这里可以通过Marcus电子转移理论进行简单的探讨39,首先考虑一个简单的反应过程:
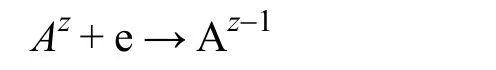
如图4a所示,假设反应物种的自由能与反应坐标成抛物线关系,那么根据简单的数学推导可以得出:

图4 反应物与产物的自由能与反应坐标的关系示意图Fig. 4 Plots of the free energy G vs. the reaction coordinate.



因此可以看出,对体系施加一定方向的过电位,可以降低反应的活化能,从而有利于反应向该方向进行。值得注意的是,并非过电位越大,反应速率就会越快,根据公式(6),活化能与过电位成抛物线关系,因此反应速率与驱动力之间存在火山型曲线,如图5所示。因此,受到动力学反应速率的限制,即使反应物处于电化学极化状态,电化学反应也有可能不会发生或仅仅以缓慢速率进行。另外,反应物与产物构型(例如配位环境、自旋状态等)差异越大,反应的活化能越高。那么我们就可以据此设计纳米晶的结构,通过优化调控过渡金属的配位环境等方式,来提高金属氧化、溶解的反应活化能,从而提高过渡金属在催化剂中的稳定性。
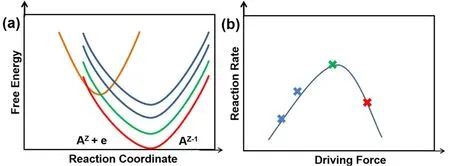
图5 反应速率与驱动力之间的火山型曲线示意图Fig. 5 Volcano relationship between reaction rate and driving force (ΔG0).
此外,由于溶解首先发生在表面,内部过渡金属的腐蚀可能需要经历一个扩散过程,即内部原子逐渐迁移到表面的过程。根据金属材料方面的知识以及菲克第一定律可知,影响物质扩散的因素有浓度梯度以及扩散系数。其中对于空位型扩散来说,扩散系数的表达式如下40,41:

其中,DA代表某个温度下A原子的扩散系数,D0代表本征扩散系数,Q为扩散活化能,ΔHm以及ΔHV分布代表迁移势垒(移动一个原子到紧邻空位所需要的能量)以及空位形成能(移除原子留下空位所需要的能量)。那么我们可以通过提高原子的迁移势垒、空位形成能等方式降低原子的扩散系数,减缓原子迁移到表面的动力学过程,从而降低原子的溶解腐蚀。Schiøtz等42采用DFT计算,报道了合金能以及迁移势垒的内在关系。根据作者的计算细节,他们提到的合金能与本文上面提到的形成焓是同一个含义。他们发现与合金能(形成焓)存在负相关关系,即合金能越负,过渡金属的迁移势垒越高(图6a);另外我们最近的研究工作表明,过渡金属的空位形成能与形成焓也存在负相关关系,即形成焓越负,过渡金属的空位形成能越高(图6b)43。根据上述提到的形成焓与内聚能的物理定义,两者也存在负相关关系。因此可以看出,铂基合金体系的形成焓或内聚能在一定程度上可以作为体系稳定性的描述符,这也从动力学角度阐述了铂-早期过渡金属合金可能在稳定性方面更具有优势。另外,由于化学键键能与铂基合金的形成焓、内聚能直接相关,因此对键能的调控也能从动力学上调控纳米晶的稳定性。
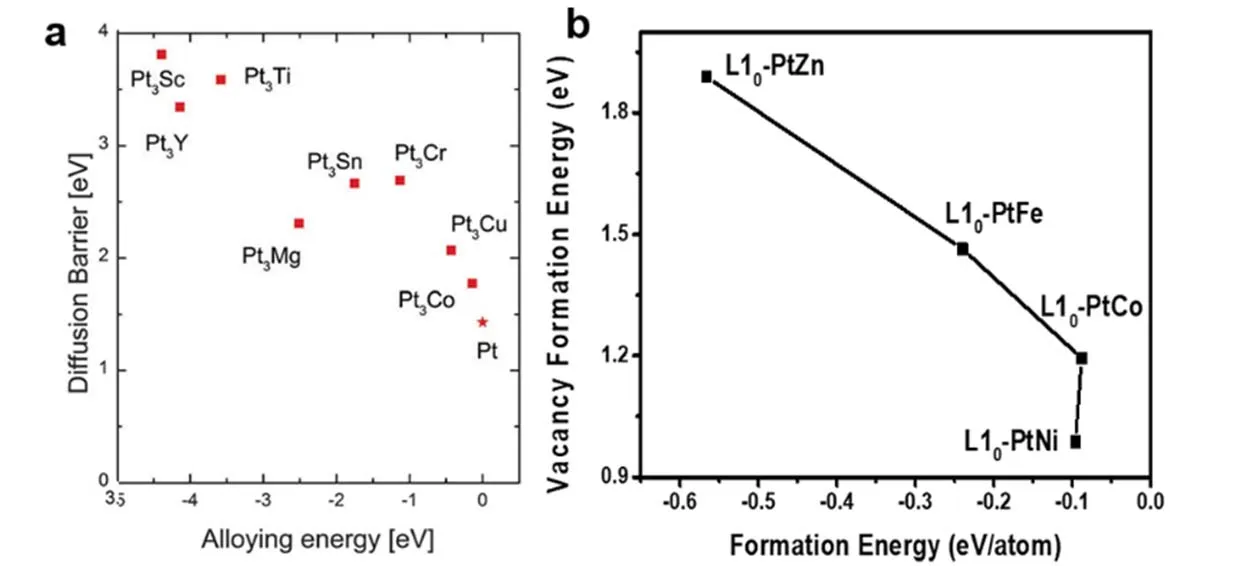
图6 (a)迁移势垒与体系合金能的关系42,(b)过渡金属的空位形成能与体系形成焓的关系43Fig. 6 (a) Relationship between diffusion barrier and alloying energy 42, (b) relationship between vacancy formation energy of transition metals and formation energy of different system 43.
3 提升稳定性的策略与方法
3.1 掺杂
大量的理论研究表明Pt-Ni催化剂,特别是Pt3Ni(111)面是活性极高的ORR催化剂13-16,对此,大量的实验工作跟进来设计结构、成分、尺寸以及形貌可控的Pt-Ni纳米晶,以实现模型预测的活性。其中,研究最多的是Pt-Ni八面体纳米粒子,目前研究者已经可以采用N,N-二甲基甲酰胺(N,NDimethylformamide,DMF)作为溶剂以及弱还原剂、或是一氧化碳封端的方法制备出成分/尺寸可控、粒径均一的Pt-Ni八面体纳米粒子44-48。这些Pt-Ni八面体纳米粒子表现出优异的ORR活性,例如Cui等46采用溶剂热方法制备的Pt-Ni八面体达到了1.45 A·mg-1(单位质量Pt,下同)的氧还原质量活性,而Choi等45制备的催化剂更进一步实现了3.3 A·mg-1的质量活性,是当时的一个最高纪录。尽管Pt-Ni纳米八面体具有优异的ORR活性,其长效稳定性仍存在一定的问题,例如上述Choi等45报道的催化剂,经过5000圈加速稳定性测试之后,Pt3Ni纳米八面体的质量活性下降了约40%。因此,如何进一步设计纳米晶的结构,从而提高催化剂的稳定性,则是另外一个较为重要的研究方向。
在Pt-Ni二元纳米晶的基础上,掺杂少量第三种金属,或许可以进一步调控纳米晶局部的配位环境以及应力分布,从而进一步优化催化剂的活性,也可能改善催化剂的稳定性。Huang等49在Pt-Ni八面体的基础上,通过羰基化合物的分解引入多种元素,包括V、Cr、Mo、W等,他们发现:Mo-Pt3Ni催化剂实现了6.98 A·mg-1的质量活性以及10.3 mA·cm-2的面积比活性,同时在8000圈循环之后,质量活性仅仅衰减5.5%,如图7a所示。DFT理论计算表明,Mo的掺入能够诱导产生较强的Mo―Pt以及Mo―Ni键,从而稳定Pt、Ni并降低元素的溶解,提高催化剂的稳定性。在随后的一篇报道中,同一个课题组的研究者采用同步辐射研究了Mo的具体作用50。他们结合分子动力学模拟以及原位同步辐射研究发现,Mo元素主要以氧化态形式存在,并优先占据边界、顶点等低配位位点(图7b),Mo的存在能够稳定近邻的Pt原子,降低紧邻Pt原子的迁移,从而稳定纳米晶的八面体形貌;而纳米晶内部的Ni则可以被表面的Pt壳层保护,阻止Ni被刻蚀、流失,这两者能够保护Pt-Ni(111)晶面,从而提高催化剂的稳定性。
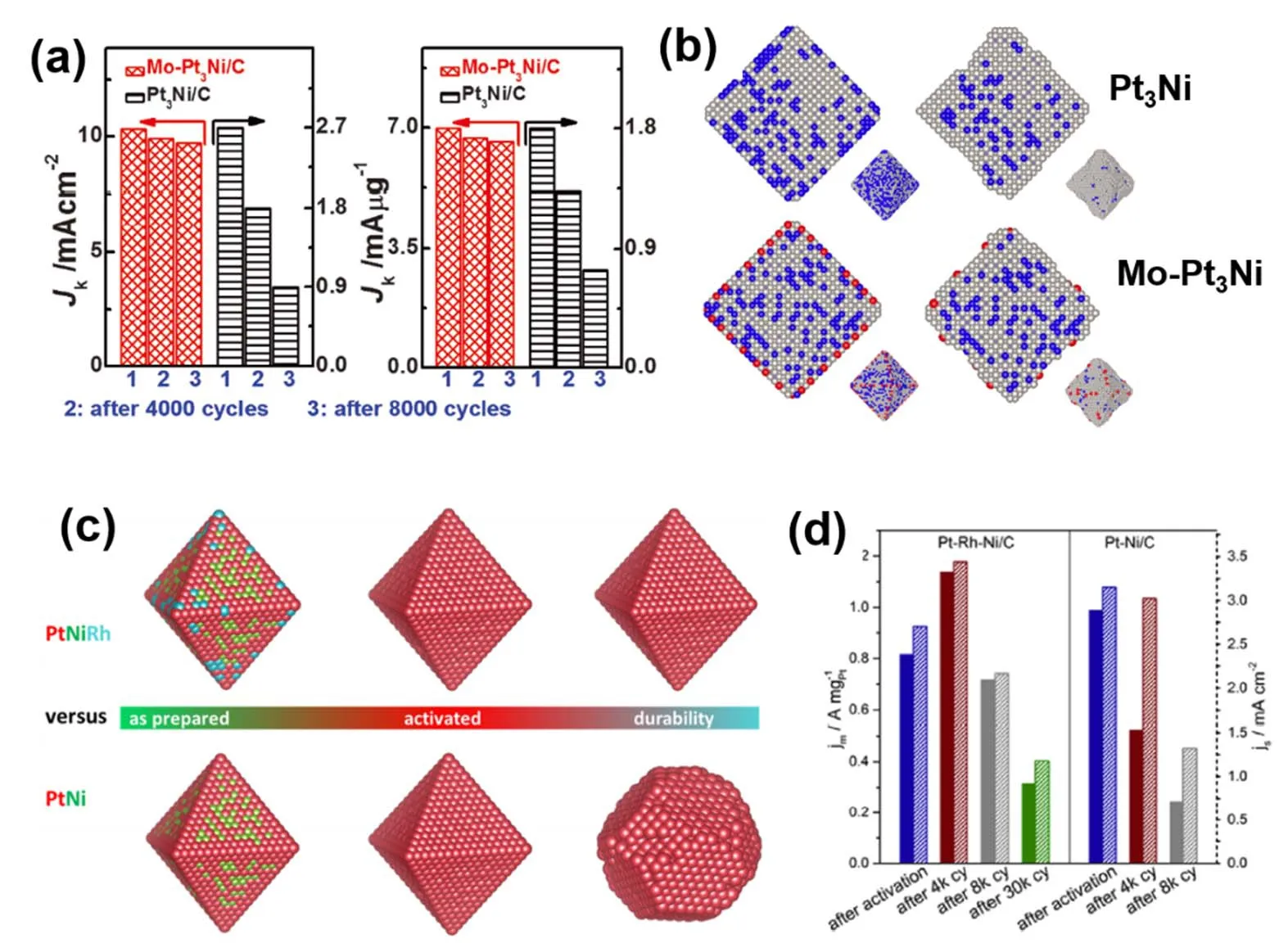
图7 (a) Mo-Pt3Ni以及Pt3Ni纳米八面体循环前后的氧还原活性49,(b)分子动力学模拟的元素分布50,(c) Rh-PtNi以及PtNi纳米八面体的形貌演变,(d) Rh-PtNi以及PtNi纳米八面体循环前后的氧还原活性51Fig.7 (a) ORR activity of Mo-Pt3Ni and Pt3Ni octahedrons before and after potential cycles 49; (b) elemental distribution of Mo, Ni and Pt obtained by kinetic Monte Carlo (KMC) simulations 50; (c) morphology evolution of Rh-PtNi and PtNi octahedrons; (d) ORR activity of Rh-PtNi and PtNi octahedrons before and after potential cycles 51.
Beermann等51认为纳米晶的性能衰减主要源于八面体形貌的坍塌,而并非Ni元素的溶解流失。他们在Pt-Ni纳米八面体的基础上,引入了少量的铑(Rh)来改善催化剂的稳定性。电化学测试结果表明,尽管Pt-Rh-Ni纳米八面体的初始ORR活性稍微低于Pt-Ni纳米八面体,但是其稳定性大大提高:稳定性测试循环4000圈之后,Pt-Rh-Ni纳米八面体的ORR质量活性从0.82 A·mg-1上升至1.14 A·mg-1,进一步循环至8000圈,Pt-Rh-Ni纳米八面体仍然保持0.72 A·mg-1,而相比之下,Pt-Ni纳米八面体循环8000圈后活性下降至0.12 A·mg-1。透射电子显微镜结果表明,Rh主要存在与纳米晶的表面,能够抑制Pt原子的迁移,因此在电化学循环的过程中,纳米晶的八面体结构能够被很好的稳定,即高活性的Pt-Ni(111)面能够在电化学循环过程中稳定存在,从而表现出优异的ORR稳定性,如图9c,d所示。相反,没有Rh掺入的纳米晶,仅仅在4000圈循环过后,八面体的形貌就已经消失,导致性能的急剧下降,如图9D所示。除了Rh元素外,类似的还有Ga52、Cu53、Fe54等,这些元素的掺入均在一定程度上保持八面体的形貌从而提高了Pt-Ni催化剂的ORR稳定性。
金元素的掺杂也引起了研究者广泛的兴趣。2007年,Adzic等55发现,在Pt纳米粒子周围修饰少量的金团簇,能够大大提高Pt纳米粒子的稳定性,即使在0.6-1.1 V (相对于可逆氢电极,RHE)的范围内循环30000圈,催化性能和电化学活性面积(ECSA)几乎没什么变化,其内在原因在于,金团簇的引入能够提高Pt的氧化电势,从而降低Pt的溶解和迁移,提高材料的稳定性;另一个可能的原因则是,Au原子可能会扩散到边、台阶等低配位的地方,阻碍了这些位点与溶液的反应,从而降低了Pt的溶解速率。随后,许多的工作也报道了Au掺杂对催化剂稳定性提升的现象,例如Au-Pt3Ni纳米线56、Au-PtCu纳米粒子57以及Au-PtCo纳米粒子58等,Au掺杂在提升催化剂稳定性方面表现出较有希望的前景。
3.2 原子排布调控
目前许多的研究主要集中在无序固溶体(无序结构),两种原子在晶体内部随机分布。除了无序固溶体之外,二元金属还存在一种热力学更稳定的有序相,即金属间化合物(有序结构),指由两个或更多的金属组元或类金属组元按比例组成具有金属特性、同时具有长程有序晶体结构的化合物。以Pt-Fe为例,Pt-Fe合金固溶体的晶体结构为面心立方结构(fcc),Pt/Fe随机分布在各个位点上;而形成Pt-Fe金属间化合物之后,Fe以及Pt会沿c轴交替排列,形成四方结构(L10),如图8所示。相较于无序固溶体,有序相具有更负的形成焓以及更强的Pt-M键能,根据第二章的分析,有序相具有更高的催化剂稳定性。
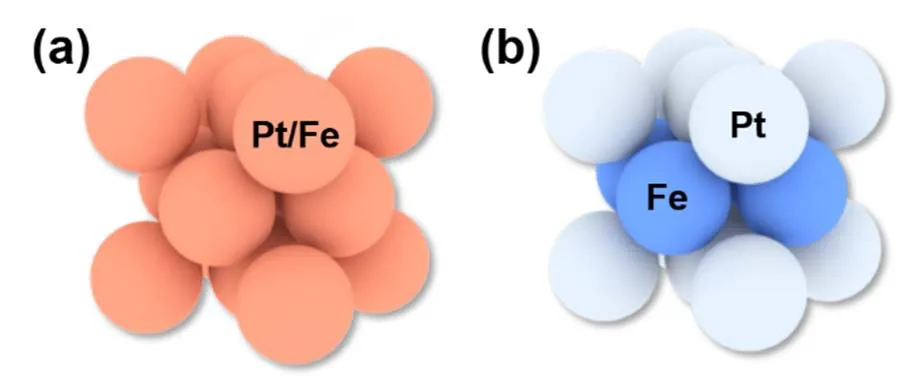
图8 无序面心立方(a)以及有序面心四方(b)结构示意图Fig. 8 Schematic illustrations of disordered fcc structure(a), and ordered L10 structure (b).
Pt-Fe以及Pt-Co金属间化合物具有优异的磁学性能,早期已经被用于磁性材料,例如信息储存等59,60。但制备有序结构通常需要高温退火(>600 °C),此时纳米粒子容易发生团聚,导致催化剂活性的下降。因此,我们需要采取一些方法保护纳米颗粒,防止其团聚而导致不良的结果。得益于材料制备技术的发展,现在研究者已经可以通过在纳米粒表面包覆氧化物61-68、碳69-71以及其他热稳定物质72,73来防止纳米粒团聚的现象。例如Chung等69在6 nm的PtFe纳米粒子表面包覆聚多巴胺,在退火过程中,多巴胺原位碳化,形成一层氮掺杂碳保护层来保护PtFe纳米粒子,同时PtFe纳米粒子自身发生相变过程,形成L10-PtFe纳米粒子(图9a)。相对于无序PtFe纳米粒子以及商业化Pt/C,L10-PtFe纳米粒子具有更好的ORR活性,质量活性达到1.6 A·mg-1。该催化剂进一步应用于燃料电池,得益于碳层的保护及独特的有序结构,该催化剂经过100 h稳定性测试之后,功率密度仅下降3.4%,表现出极佳的稳定性。而在相同条件下,商业化铂碳的功率密度下降27%。随后,Du等70用乙炔为碳源,制备了直径只有3 nm的L10-PtFe纳米颗粒,得益于有序结构以及较小的尺寸,该催化剂也表现出优异的ORR活性以及燃料电池性能。

图9 (a)碳包覆L10-PtFe纳米粒子的制备流程图69,(b)完全有序L10-PtFe纳米粒子的制备流程图,(c) L10-PtFe纳米粒子的ORR极化曲线,(d) L10-PtFe纳米粒子循环后的扫描透射电镜图62,(e) L10-PtFe/Pt纳米粒子的高温稳定性11Fig. 9 (a, b) Schematic illustrations of the preparation of L10-PtFe NPs 62,69; (c) ORR polarization curves of various PtFe NPs; (d) STEM image of L10-PtFe NPs after stability test 62; (e) high temperature stability of L10-PtFe/Pt NPs 11.
在退火过程中,为了提高原子的迁移能力,Li等62设计了哑铃状的PtFe-Fe3O4的结构作为前驱体来制备完全有序L10-PtFe纳米颗粒。在Ar/H2还原性气氛中,Fe3O4会被还原成Fe,形成大量的氧空位,增加原子移动的空间,从而得到完全有序的纳米颗粒,如图9b所示。从磁滞回线上看,L10-PtFe纳米颗粒的矫顽力达到33 kOe,表明结构具有极高的有序度。电化学结果表明,随着有序度的增加,催化剂的ORR性能逐渐提高,完全有序的L10-PtFe纳米颗粒ORR半波电位高达0.958 V (图9c)。同时催化剂具有极佳的稳定性,在20000圈循环后性能没有明显变化,纳米晶的有序结构也能完好保持,如图9d所示,由于Pt与Fe的原子序数不同,从扫描透射电镜中可以看出明显的亮暗差别,并且呈现明暗相间的结构。目前大部分报道的稳定性都是在室温下测试,而实际燃料电池一般在60-80 °C下运行,可能会加速催化剂活性的衰减,因此在测试催化剂的高温稳定性可能更具有实际意义。尽管完全有序的L10-PtFe纳米颗粒在室温下具有优异的稳定性,但在60 °C下,其性能衰减较多11。对此,Li等11对上述制备的L10-Pt-Fe纳米粒子进行酸洗、后退火预处理,在纳米晶的表面形成完整的Pt-skin结构。该Pt-skin结构的存在能够进一步保护纳米晶内部的Fe原子,从而大大提高了催化剂的稳定性,即使在60 °C下循环10000圈也没有明显的活性下降,如图9e所示。Zhang等74深入研究了L10-PtFe相对于无序PtFe纳米晶活性提高的原因。他们通过理论计算发现,当无序PtFe纳米晶向有序L10-PtFe相变之后,沿[100]以及[010]方向的晶格常数会变大,而沿[001]方向的晶格常数会减小,两者共同作用会导致表面Pt原子受到的压缩应力减小,优化了Pt-O的结合能,因此有序L10-PtFe纳米晶具有更好的ORR催化活性。
Pt-Co体系最近也吸引了较多的关注。Wang等75将Pt、Co前驱体以及炭黑进行简单混合,随后在氩氢气氛下,分别在400 °C以及700 °C下退火,即可得到无序Pt-Co纳米粒子以及有序Pt3Co纳米粒子。其中Pt3Co的晶体结构与L10-PtFe的有所不同,其晶体结构为L12(立方晶系),Co主要占据晶胞的八个顶点位置,Pt占据6个面心位置。电化学测试结果表明,L12-Pt3Co催化剂不仅具有优异的ORR活性,同时也表现出更好的稳定性。最近,Li等76采用坚果状的PtCo纳米颗粒为前驱体,在650 °C下退火制备了有序L10-PtCo纳米颗粒。得益于纳米粒独特的坚果状形貌,纳米晶不仅在退火过程中没有发生团聚,同时原子的迁移能力也大大提高,得到了有序度较高的L10-PtCo纳米颗粒。与上述L10-PtFe类似,作者结合酸洗以及后退火处理,在L10-PtCo纳米颗粒表面形成了约2-3原子层厚的Pt-skin结构,如图10a所示。得益于Pt-skin结构以及有序的L10-PtCo结构,该催化剂在半电池中具有优异的ORR活性以及高温稳定性,在60 °C下循环30000圈仍保持了80%的活性。重要的是,该催化剂也成功应用于燃料电池中,也表现出优异的稳定性,电池性能在30000圈循环(0.6-0.95 V,方波)之后几乎没有衰减(图10b),其燃料电池活性以及稳定性均达到美国能源部设定的技术指标。最近,Liang等77采用Pt/CoO核/壳结构作为前驱体,制备了直径为3 nm的超细L10-PtCo纳米晶作为高效的燃料电池阴极催化剂。他们发现,在L10-PtCo中掺入~5%的W元素能够一方面进一步优化Pt壳层的表面应力,提高催化活性;另一方,W掺杂能够降低纳米晶的表面能,协同有序结构一起提高催化剂的稳定性(图10c)。在燃料电池测试中,该催化剂在50000圈循环之后性能衰减< 30% (图10d)。随后该研究小组43设计了一种Pt纳米粒子镶嵌在ZnO模板中的复合结构作为前驱体,制备了超细的L10-PtZn/Pt纳米粒子。优于L10-PtZn核与Pt壳层晶型以及晶格常数不同,PtZn核与Pt壳层会产生沿<110>方向的拉伸应力以及沿<011>方向的压缩应力,该双轴应力协同调控Pt壳层的应力分布,提升了催化剂的ORR活性。而有序结构则提升Zn原子的空位形成能,进而提升催化剂的稳定性。最终,该L10-PtZn/Pt催化剂在PEMFC测试中表现出良好的性能,功率密度达到2.0 W·cm-2,而循环30000圈后活性衰减小于20%。
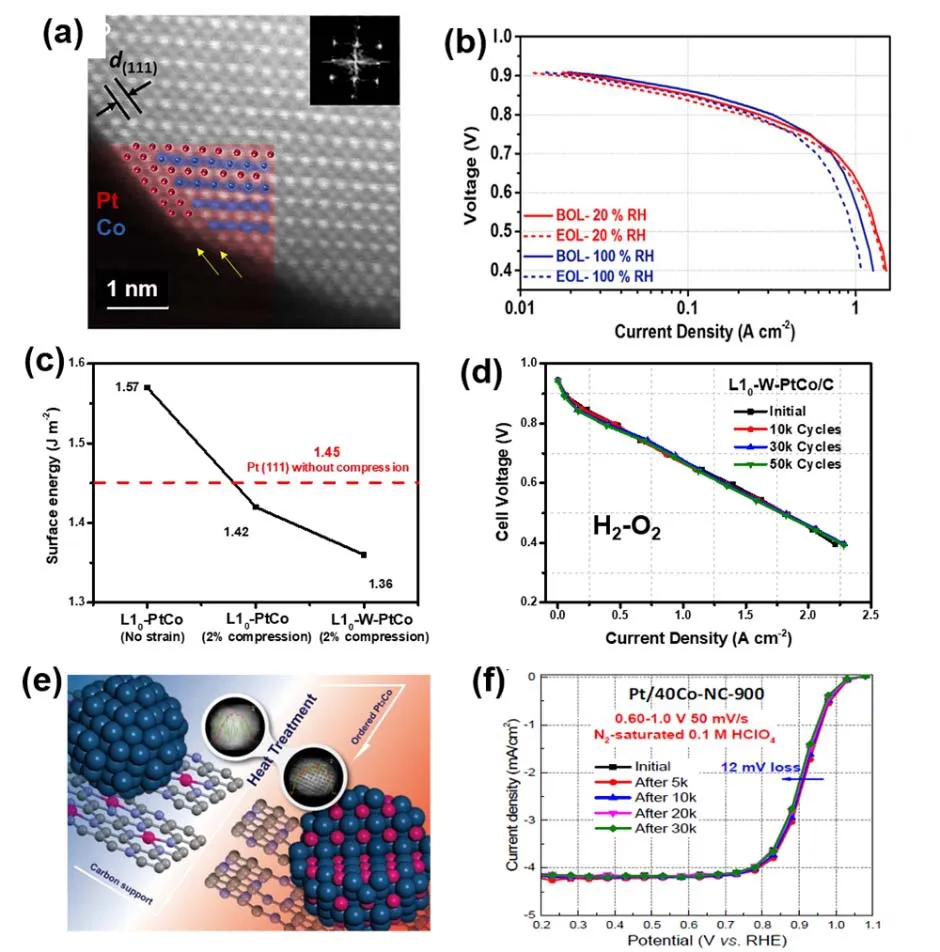
图10 (a) L10-PtCo/Pt纳米粒子的描透射电镜图,(b) L10-PtCo/Pt纳米粒子在燃料电池中的性能76,(c)不同催化剂的表面能,(d) L10-W-PtCo纳米粒子在燃料电池中的性能77,(e) N-C负载L12-Pt3Co纳米粒子的制备流程图,(f) N-C负载L12-Pt3Co纳米粒子的ORR稳定性78Fig. 10 (a) STEM image of L10-PtCo/Pt, (b) fuel cell performance of L10-PtCo/Pt 76; (c) surface energy of different catalysts,(d) fuel cell performance of L10-W-PtCo 77; (e) schematic illustration of the preparation of L12-Pt3Co,(f) ORR polarization of L12-Pt3Co before and after potential cycling 78.
目前大部分有序结构都是通过首先制备PtM合金纳米晶,随后通过高温退火的方法得到PtM有序结构纳米晶,而Wang等78提出了一种新的制备L12-Pt3Co纳米颗粒的方法。他们首先用ZIF-67制备了原子级分散的Co-N-C材料,在此基础上原位制备了Pt纳米颗粒,随后在高温退火过程中,原子级分散的Co会脱离碳基底,进入Pt晶格,进而发生相转化生成L12-Pt3Co纳米颗粒;与此同时,L12-Pt3Co纳米颗粒与N-C基底之间也会形成较强的相互作用,协同提高催化剂的ORR活性和稳定性,如图10e所示。电化学测试结果表明,L12-Pt3Co纳米颗粒具有较好的ORR活性,在0.85 V下面积比活性达到5.1 mA·cm-2;而且催化剂的稳定性也比较优异,在30000圈循环后半波电位仅仅下降12 mV,而且电镜结果表明催化剂的结构在循环之后也较完整的保留(图10f)。
3.3 物理、化学限域作用
通过纳米晶限域、提高纳米晶与与载体的作用面积/强度等方法,可以降低纳米晶团聚的趋势,减缓纳米晶的溶解从而提高催化剂的稳定性。例如Khlobystov等79采用了一种具有多级台阶的碳纳米管作为保护层以及载体,抑制了Pt纳米粒子在电催化过程中的团聚现象,提高Pt纳米粒子的稳定性,在循环50000圈后,催化剂的粒径分布、ECSA以及氧还原活性均具有较好的保持。同样,Sun等80通过原子层沉积的方法,在Pt纳米粒子周围沉积了一圈ZrO2作为物理限域的障碍,防止Pt纳米粒子在电化学测试中发生迁移、团聚,从而提高催化剂的稳定性。Liu等37设计了一种具有原子级分散的Fe-N-C载体负载Pt-Fe纳米粒子,作为高稳定的氧还原催化剂,其中载体中的Fe能够与Pt合金化,提高纳米粒子的ORR活性,而N掺杂缺陷位以及Fe-N4位点对纳米粒子进行锚定,并诱导金属载体相互作用,提高催化剂的稳定性。类似的材料设计还可以进一步拓展到Pt-Co体系35,36。
由于一维纳米线具有独特的各项异性的特点,纳米线结构在ORR催化过程中具有较多的优势。(1)纳米线通常具有光滑的表面,较少的缺陷,具有较大的电化学活性面积,能够提高催化剂的活性;(2)纳米线一维的形貌有助于电子以及反应物种的传输,加快反应进程;(3)纳米线与载体的接触面积更大,可以有效地避免催化剂在电化学测试过程中的溶解、团聚等现象,从而提高催化剂的稳定性。因此,铂基纳米线催化剂也得到了研究者广泛的关注。最近,Jiang等81报道了一种仅4-5个原子层厚的亚纳米级别的纳米线催化剂,如图11a所示。他们采用十六烷基三甲基氯化铵作为表面活性剂,羰基钼、葡萄糖作为还原剂。作者做了大量的对比试验,探究了各个反应物对纳米晶形貌的影响,发现过渡金属离子的引入能够减小纳米线的直径,而十六烷基三甲基氯化铵以及羰基则是纳米线形成的关键。该方法具有一定的普适性,能够制备纯Pt、PtCo、PtNi以及PtNiCo等纳米线。由于纳米线超细的直径,催化剂具有较高的ECSA,约80 m2·g-1;其中PtNiCo三元纳米线具有最好的ORR活性,质量活性达到4.2 A·g-1,而且在30000圈循环后没有明显的衰减,如图11b,c所示。进一步的理论计算研究表明,PtNiCo纳米线暴露了大量的(111)晶面,该晶面具有适中的Pt-O结合能,因此具有较高的ORR活性。由于该方法具有一定普适性,随后也有许多相关的报道82,83。Huang等84进一步在Pt纳米线的基础上,掺入少量的金属Rh,实现了催化剂较高的稳定性。Rh-Pt纳米线不仅具有1.41 A·mg-1的质量活性,而且在循环10000圈之后仍保持了90.8%氧还原活性,表现出较好的稳定性,如图11d所示。DFT计算结果表明,Rh的掺入能够显著提高表层Pt原子的空位形成能,降低Pt原子的溶解,从而保持催化剂的氧还原活性,如图11e所示。Rh的掺杂作用随后也进一步拓展到PtNiRh三元纳米线中85。相比于PtNi二元纳米线,该PtNiRh三元纳米线具有更高的催化稳定性,循环10000圈后仅衰减12.8%。

图11 (a) PtNi超细纳米线的透射电镜图1,(b、c) PtNiCo超细纳米线的ORR活性81,(d) Rh-Pt超细纳米线的ORR活性,(e) Rh掺杂对Pt空位形成能的影响84Fig. 11 (a) STEM image of PtNi nanowires (NWs) 1; (b, c) ORR activity of PtNiCo NWs before and after potential cycles 81;(d) ORR activity of Rh-Pt NWs before and after potential cycles, (e) vacancy formation energy of different catalysts 84.
3.4 催化剂载体设计
除了活性中心,即铂基纳米晶的稳定性外,催化剂载体对于催化剂整体的稳定性也至关重要。催化剂载体能够稳定、分散纳米晶,提高纳米晶的利用率,而且能够提高物质传输、电子传输效率,提升催化剂的活性。然而,目前催化剂常用的碳载体,包括XC-72和Ketjen黑等无定型碳,稳定性仍然存在较大问题。
质子交换膜燃料电池正常运行条件下,开路电压为1.0 V左右,而在燃料电池开关机条件下,阳极可能存在少量的氧气或空气,空气会在阳极的铂催化剂表面发生氧还原反应,导致阴极电极电势上升至1.5-2.0 V86,如图12a所示。在这种高电位条件下,碳基底极易发生氧化,导致纳米晶脱落、团聚以及载体导电性的降低,最终导致催化剂失活。因此,设计高稳定的催化剂载体,对于提高催化剂稳定性,具有十分重要的实用意义。
为了解决碳载体稳定性不足的问题,研究者设计一些非碳基的载体,包括铟掺杂的二氧化锡87、钽掺杂的二氧化锡88、以及二氧化钛89,90等氧化物,来提高催化剂在高电位下的稳定性。Rozière等88设计了一种五价金属掺杂(钽、锑、铌)的二氧化锡纳米纤维,即Ta/SnO2、Sb/SnO以及Nb/SnO2作为载体负载Pt纳米粒子,他们发现,1% (原子分数)的Ta掺杂量、7% (质量分数)的Pt负载量的催化剂具有最高的氧还原活性,质量活性达到0.465 A·mg-1,明显优于商业化Pt/C。该催化剂良好的氧还原活性可以归因于金属-载体强相互作用,调控了Pt纳米粒子的电子结构,优化了Pt-O吸附能,如图12b所示。更重要的是,该催化剂在1.0-1.6 V电位区间内循环6000次后,其氧还原极化曲线基本没有变化(图12c),且ECSA以及质量活性仍保持70%以上,远优于Pt/XC-72等碳载铂催化剂。其优于的稳定性主要起源于SnO2自身良好的电化学稳定性。
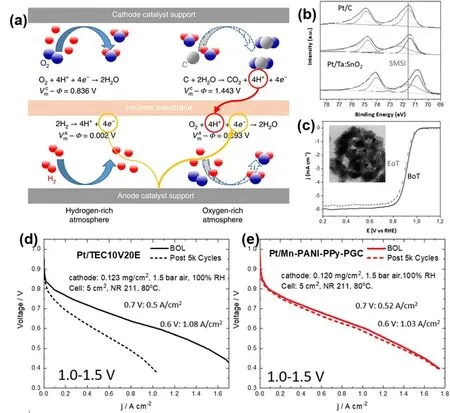
图12 (a)质子交换膜燃料电池开关机条件下阴阳极电势变化示意图86,(b) Pt/C以及Pt/Ta:SnO2的XPS图(c)高电压循环前后Pt/Ta:SnO2的氧还原计划曲线图88,(d) Pt/C以及(e) Pt/Mn-PANI-PPy-PGC催化剂循环前后的燃料电池极化曲线91Fig. 12 (a) A schematic of anodic and cathodic electrochemical processes occurring during SU/SD of PEMFCs 86;(b) XPS results of Pt/C and Pt/Ta:SnO2, (c) ORR polarization of Pt/Ta:SnO2 before and after high potential cycles 88;H2-air fuel cell polarization curves of Pt/C (d), and Pt/Mn-PANI-PPy-PGC (e) before and after high potential cycles 91.
提高碳基底的石墨化程度,也是提高载体高电位稳定性的一种有效方法,但目前石墨化碳存在的主要问题在于,其孔隙率、比表面较低,不利于物质传输以及Pt纳米晶的负载91。针对这一问题,Wu等91通过聚合物凝胶热解的方法,制备了一种三维多孔、高度石墨化的碳载体(Mn-PANI-PPy-PGC)。其中,聚合物凝胶前驱体具有三维交联骨架,能够在热解过程中产生多孔结构,有利于Pt纳米晶的负载以及后续催化过程中的物质传输。而前驱体中的锰(Mn)元素能够在热解过程催化碳载体的石墨化,提高石墨化程度。作者在该研究中系统比较了不同热解温度(900-1100 °C)、不同催化元素(Fe、Co、Mn、Ni)对碳载体石墨化程度以及催化剂稳定性的影响。结果表明,热解温度为1100 °C、且选用Mn元素催化得到的碳载体石墨化程度最高,随着石墨化程度的提高,催化剂的稳定性也逐渐提高。此外,得益于载体具有较高的孔隙率以及强金属-载体相互作用,该催化剂也表现出优于商业化Pt/C的氧还原性能。而且更重要的是,该催化剂不仅仅在半电池中具有良好的ORR活性以及高电压稳定性,在实际质子交换膜燃料电池测试中也表现出优异的性能:在0.6 V下电流密度达到1.03 A cm-2,与商业化Pt/C接近(1.08 A cm-2);而在1.0-1.5 V电位区间循环5000圈后,电流密度几乎没有衰减,如图12d,e所示。且在线气相色谱检测结果也证明,该催化剂的碳氧化程度远低于商用的碳载体。
4 总结与展望
目前PEMFC阴极铂基催化剂受到了研究者广泛的关注,高效、稳定的铂基催化剂的设计与开发,依然是燃料电池大规模应用的关键。基于d-带重心等理论模型的指导和纳米合成技术的发展,研究者目前已经可以精确地设计和合成尺寸、形貌、和结构可控的纳米晶催化剂,极大地提高了催化剂的氧还原活性,取得了十分突出的成果。另一方面,研究者们也提出了若干策略来提高催化剂的稳定性,包括过渡金属掺杂、原子排布有序化调控以及物理限域等方法,也取得了一定的成效。然而,相比于活性,催化剂稳定性研究还存在较大的拓展空间。
(1)稳定性的理论研究仍然缺乏统一的理论,不同文献采用的计算模型、方法可能有所不同,导致最后的计算结果也可能存在区别。如何搭建合理、普适的模型,寻找合适的稳定性描述符,可能是未来理论研究的一个重要方向。
(2)催化剂结构演变的表征仍存在问题,电催化过程涉及到气相、液相以及固相等多相界面,催化剂结构演变的表征可能比较困难。然而,实时追踪催化剂结构的演变,对于更深入的理解催化剂稳定性衰减的机理以及推动理论模型的搭建十分重要。因此,如何构筑表征-测试联用仪器,实时追踪结构的变化,可能是未来实验上的一个研究方向。
(3)半电池与燃料电池测试条件的区别,可能会导致催化剂活性、稳定性表达的区别。目前大部分催化剂均在半电池进行性能的表征,而燃料电池的运行温度、膜电极结构等与半电池测试具有较大的区别,这些因素可能会加速催化剂的衰减,导致催化剂在燃料电池中的稳定性较差。因此,催化剂需要尽可能地在燃料电池中评估其活性以及稳定性。
总之,研究者们需要针对PEMFC阴极催化剂稳定性的问题,进一步地深入研究,设计高效、稳定的PEMFC阴极催化剂催化剂,推动PEMFC的商业化进程。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Enhanced Performance and Durability of High-Temperature Polymer Electrolyte Membrane Fuel Cell by Incorporating Covalent Organic Framework into Catalyst Layer
- Formic Acid Electro-Oxidation Catalyzed by PdNi/Graphene Aerogel
- 金属卟啉修饰的多孔聚苯胺基氧还原电催化剂
- 有序金属间化合物电催化剂在燃料电池中的应用进展
- 高温聚合物电解质膜燃料电池膜电极中磷酸分布及调控策略研究进展
- Recent Progress in Proton-Exchange Membrane Fuel Cells Based on Metal-Nitrogen-Carbon Catalysts