Alagille综合征1例报告
2021-09-25宋文艳郑素军
汤 珊, 白 丽, 宋文艳, 梁 晨, 白 洁, 郑素军
1 首都医科大学附属北京佑安医院 a.肝病中心一科, b.肝病中心四科, c.影像科, 北京 100069;
2 肝衰竭与人工肝治疗研究北京市重点实验室, 北京 100069
Alagille综合征(Alagille syndrome,ALGS)是一种多数由JAG1(Jagged canonical Notch ligand 1)基因突变或缺失引起的常染色体显性遗传病[1]。Alagille综合征的典型临床特征有:肝内小叶间胆管数量减少或缺乏,慢性胆汁淤积;心脏杂音;角膜后胚胎环;蝴蝶椎体;特殊面容,如前额突出,眼眶深陷,眼距增宽等[2]。早期,根据婴儿胆汁淤积症诊断的近似估计,该病的发病频率为1/70 000。 然而,随着分子诊断技术的进展,真实发生率可能接近1/30 000[3]。ALGS的临床表现形式多样,即使在同一家族中,也可能有所不同,给诊断带来一定困难。现报道本院诊治的1例ALGS病例,供临床参考。
1 病例资料
患者男性,26岁,2020年5月于体检时发现肝脾肿大,伴皮肤轻度瘙痒,无乏力、腹胀、恶心、呕吐等不适,曾于外院诊断未明而来本院。既往史:既往无规律体检,19年前因室间隔缺损行修补手术治疗,术后恢复良好;血糖升高数月,未诊治;无病毒性肝炎病史,无吸烟、饮酒史;否认近期用过其他药物、滋补品。家族史:父母未行肝功能、基因检测等检查,否认类似疾病家族史。本次入院查体:生命体征平稳,发育正常,前额突出,眼球深陷伴眼距中度增宽,尖下巴,营养良好。牙齿正常,皮肤、巩膜无明显黄染,K-F环阴性,皮肤无皮疹、出血点及瘀斑,胸正中线皮肤可见20 cm手术瘢痕。心、肺听诊未闻及异常。腹部平坦,无压痛和反跳痛,未触及包块,肝脏肋下3 cm,剑突下2.5 cm,质软,脾脏肋下未触及,Murphy征阴性,移动性浊音阴性,肠鸣音正常。双下肢无水肿。四肢脊柱均无明显畸形。
实验室检查显示,ALT、AST、TBil、DBil、TBA、ALP、GGT均明显升高,PLT降低(表1)。血氨、凝血功能、肝纤维化四项、肿瘤标志物、尿常规、粪便常规、心肌酶、肾功能、铜蓝蛋白均未见明显异常,HBsAg阴性、抗-HBs阳性、HBeAg阴性、抗-HBe阴性、抗-HBc阴性、丙型肝炎抗体阴性、HIV抗体阴性、梅毒螺旋体抗体阴性、自身免疫性肝病抗体阴性。上腹部CT显示(图1):肝脏各叶比例失常,右叶体积增大,呈肥大性假肿块样改变,增强扫描静脉期可见门静脉及属支管腔扩张,门静脉主干管径约1.8 cm,三支肝静脉显影,肝中静脉粗大,远端与门静脉属支似有交通,延迟期肝右叶团块影强化低于肝左叶,下腔静脉显影良好,肝内最窄处约0.9 cm,脾脏增大,胆囊移位,胆管未见扩张。减影血管造影下静脉造影显示:下腔静脉、肝右静脉、肝中静脉血流通畅。胃镜未见明显异常。肝活检病理提(图2)示:胆管消失、胆管发育不良;肝细胞慢性淤胆。

注:a,小叶间动脉可见,有多个分支,但未见伴行胆管,汇管区边缘见增生的小胆管结构,上皮排列不整、细胞轻度增生,未见明确管腔形成,提示胆管消失(箭头)、胆管发育不良(HE染色,×200);b,肝实质局部见肝细胞排列呈假腺样结构(箭头)(HE染色,×200);c、d,灶状肝细胞阳性,提示肝细胞慢性淤胆(CK7免疫组化染色,c,×40;d,×100)。

表1 临床检验结果变化
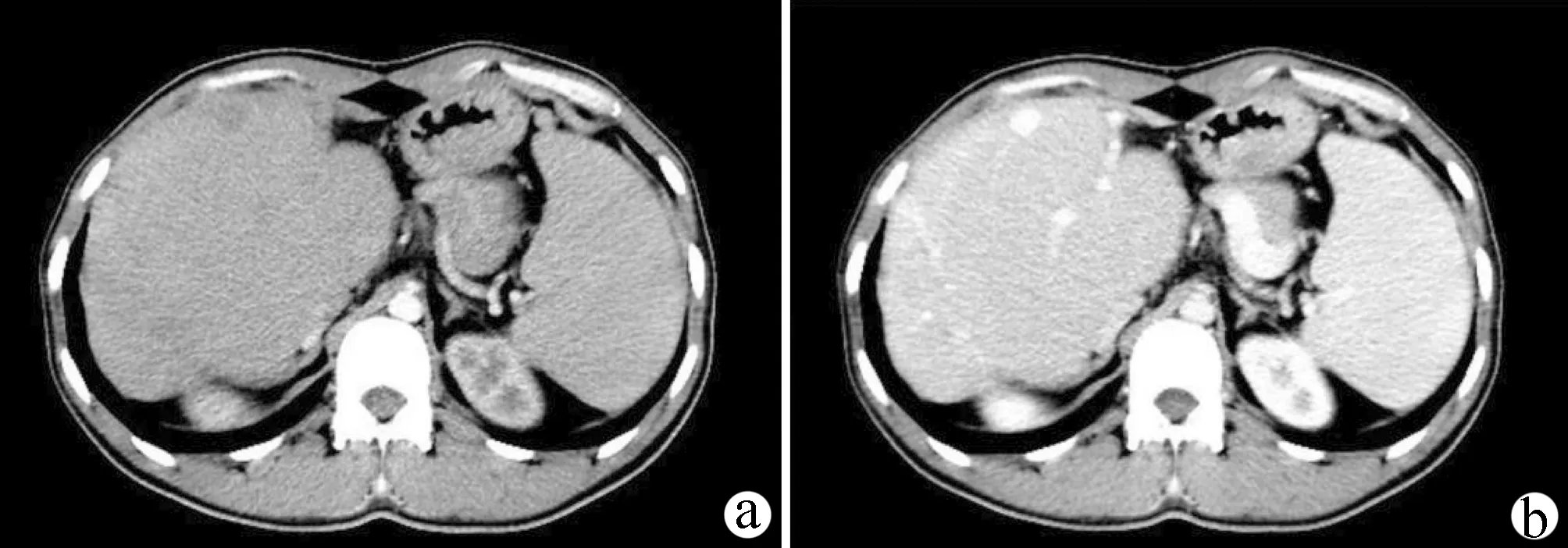
注:a,静脉期;b,动脉期。
2020年10月患者行全外显子二代基因测序并经Sanger测序验证,结果显示: JAG1基因第13外显子第1665碱基位点T突变为A,使第555氨基酸位点的酪氨酸发生无义突变,使蛋白翻译异常终止,提示Alagille综合征(图3,表2)。
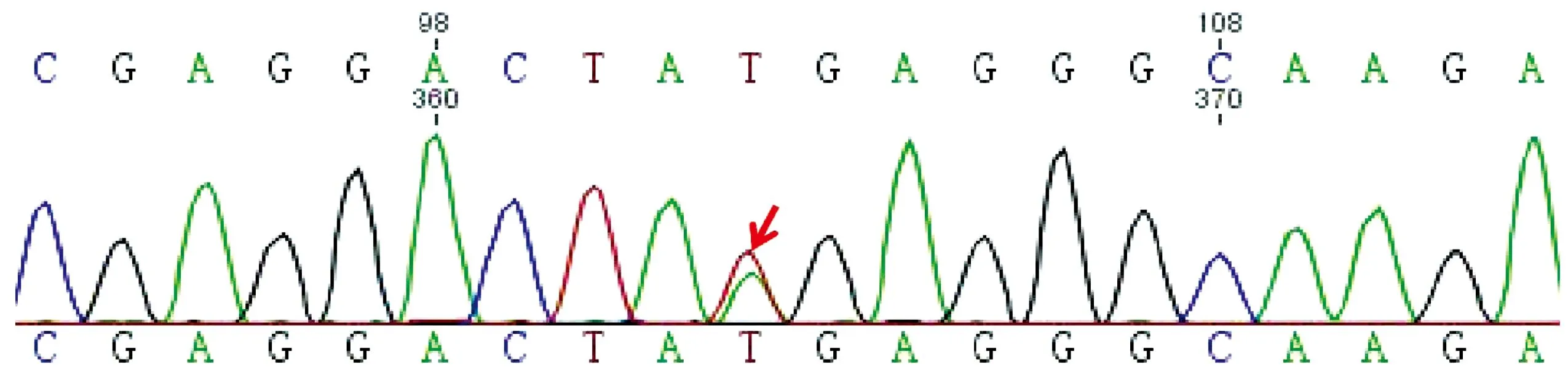
注:JAG1基因第13外显子发生杂合突变,c.1666 T>C,p.Y555X。箭头指示杂合突变。

表2 基因检测结果
综上,患者临床表现为肝脾大,化验检查ALP高达558 U/L(>3倍正常值上限),GGT高达1514U/L(>1.5倍正常值上限),存在胆汁淤积。肝活组织病理检查显示肝内小叶间胆管数量减少/消失,慢性胆汁淤积。基因检测JAG1基因第13外显子c.1666 T>C杂合突变; p.Y555X。结合患者的特殊面容,排除常见的病毒性肝炎、酒精性肝炎、自身免疫性肝炎及药物性肝炎。患者进一步完善眼科检查,未发现角膜后胚胎环。完善胸椎CT后发现患者有明显的蝶状椎骨(图4)。根据传统诊断标准[4]:患者存在JAG1基因突变,且有前额突出,眼眶深陷,眼距增宽的特殊面容、慢性胆汁淤积2种典型表现以及蝶状椎骨,可明确诊断为Alagille综合征。
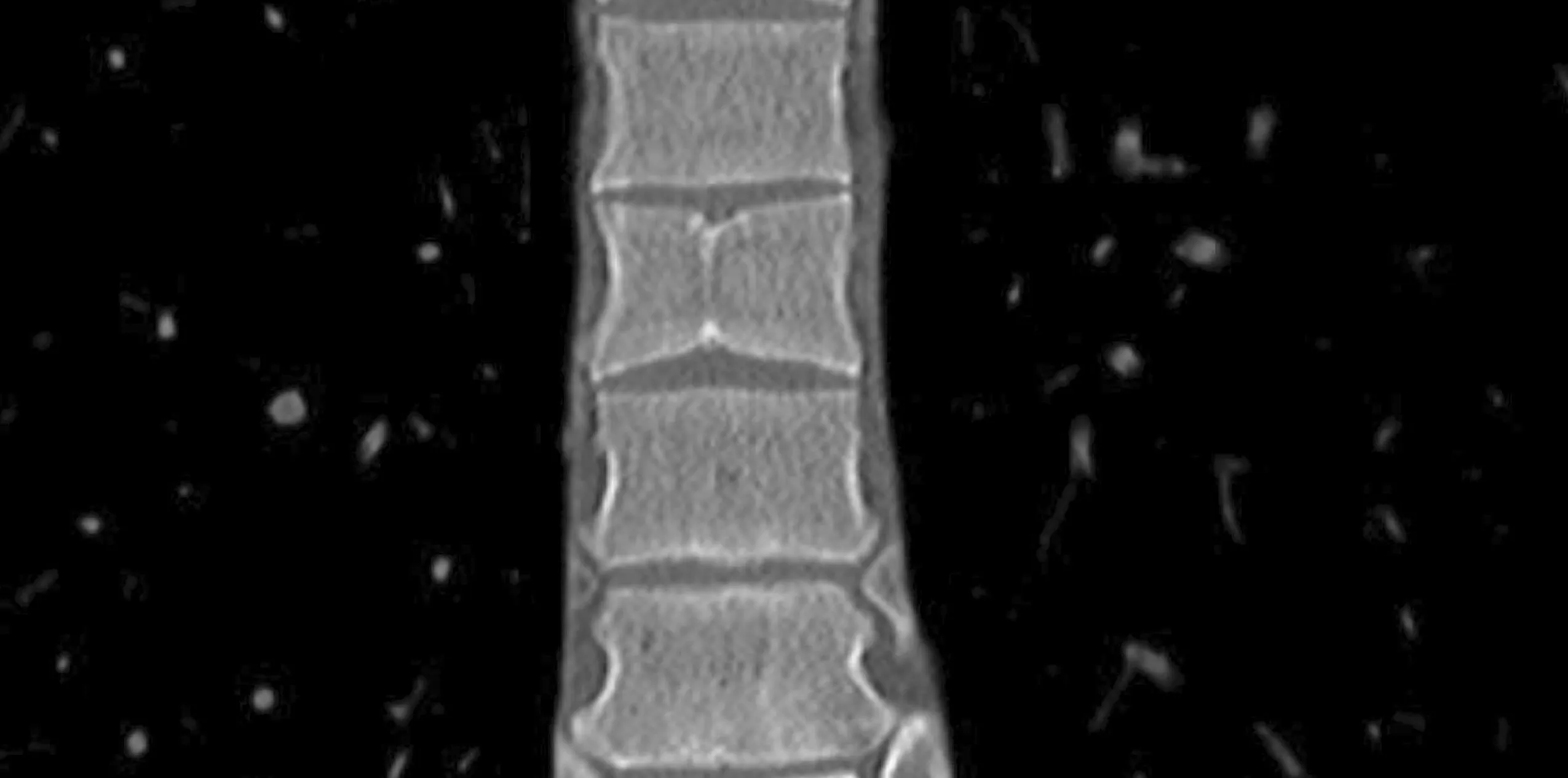
注:第7胸椎可见明显蝶状椎骨。
治疗上从2020年6月开始,主要给予熊去氧胆酸胶囊(250 mg,3次/d),并补充脂溶性维生素D3。3个月后患者瘙痒明显好转,复查肝功能显示,ALT、AST、TBil、DBil、ALP、GGT均有下降(表1)。目前患者仍在随访中。
2 讨论
ALGS是一种累及多系统的常染色体显性遗传疾病,1969年由Alagille首次报道[5]。迄今为止,全世界仅报道过500例ALGS,国内的报道则更少。94%的ALGS由编码JAGGED1的JAG1基因突变或缺失引起,约1.5%由NOTCH2(NOTCH-receptor2)基因突变导致,这2个基因都在Notch信号通路中起作用[6]。除此之外,另有4.5%未检测到基因突变。ALGS的诊断主要是通过典型的临床表现,即在肝内胆管缺失/减少的基础上具有以下5种典型表现中的3种即可以诊断:(1)慢性胆汁淤积;(2)眼部异常(角膜后胚胎环);(3)先天性心脏病(以肺动脉狭窄为主);(4)脊柱畸形(蝴蝶状椎骨);(5)特殊面容(前额突出,眼眶深陷,眼距增宽,鼻梁扁平,尖下巴等)。存在JAG1基因突变或有ALGS家族史时,合并2种典型表现即可诊断[7]。
ALGS 因小叶间胆管缺失/减少,常表现为胆汁淤积,其往往临床变异大、轻重不一,常导致漏诊、延迟诊断,甚至误诊。ALGS的诊断及鉴别诊断上应注意以下要点:(1)鉴别胆汁淤积的部位。胆汁淤积根据发生部位可分为肝内和肝外胆汁淤积两大类[8]。肝外胆汁淤积主要由胆道肿瘤、结石或狭窄引起胆道梗阻所致,MRCP等影像学检查常有助于排除诊断。本患者影像学检查胆道未发现异常,故患者胆汁淤积非肝外因素所致。(2)应进一步排除肝内胆汁淤积的常见病因。肝内胆汁淤积根据细胞学损害的部位不同可分为胆管细胞性和肝细胞性。胆管细胞性胆汁淤积的常见病因包括PBC、小胆管PSC及合并自身免疫性肝炎。血清中的自身抗体如ANA、AMA、抗 Sp100 抗体等及病理检查有助于诊断。肝细胞性胆汁淤积主要病因有病毒性肝炎、酒精、药物等,经积极询问病史或完善病毒标志物等检查有助于排除。本例患者上述检查均未发现明显异常。(3)应警惕遗传代谢性肝病等罕见病、少见病。在排除胆汁淤积的等常见原因后,特别是青年患者,应考虑遗传代谢性肝病等少见病。结合临床表现、影像、病理、基因检测等结果,进行综合判断,必要时可开展多学科会诊,有助于正确诊断。本例患者以肝脾肿大,肝功能异常起病。经检查发现患者胆系酶升高,主要表现为胆汁淤积。首先仔细询问患者病史,排除了药物性肝损伤、酒精性肝病所致的胆汁淤积性肝病。经影像学检查排除了肝外胆道梗阻这一病因。又继续完善自身抗体检查及肝组织活检,排除自身免疫性肝病。之后进一步考虑罕见的胆汁淤积性肝病,完善基因检测,发现JAG1基因突变,结合患者特殊面容及典型的蝶状椎体表现终得以确诊。
ALGS的治疗,以对症治疗为主,包括:(1)补充营养;(2)应用利胆剂如熊去氧胆酸;(3)应用降胆固醇药物如消胆胺;(4)严重瘙痒药物治疗无效时可行胆道分流手术;(5)出现继发性门静脉高压,严重胆汁淤积,难治性瘙痒等可进行肝移植手术[9]。本例患者住院与门诊随访期间主要予以熊去氧胆酸等药物保肝利胆。目前患者血清肝功能好转,但胆固醇仍高于正常水平,在继续予患者保肝利胆的同时可加用降胆固醇药物。目前患者症状改善,随访期间未出现肝外明显病变。
成人ALGS发病率低,据笔者所知,2017年首例成人ALGS才第一次被诊断[10]。本例患者儿童期未见明显黄疸及其他症状,成年以后因体检发现肝脾大就诊,最终经基因检测确诊为ALGS。但是由于患者儿童时期就诊资料不全,ALGS是成人期发现的,还是成人期发病的,目前尚不能完全确定。一方面这提示了定期体检的重要性,另一方面说明胆汁淤积的病因诊断需要拓宽思路。对于原因不明的成人胆汁淤积,应警惕遗传代谢性疾病等罕见病可能,必要时行相关基因检测明确诊断。关于此病基因型与表型的关系还待进一步研究确定。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:汤珊、白丽负责撰写论文;宋文艳提供影像学资料及分析;梁晨、白洁参与临床资料收集;白洁、郑素军负责文章修改;郑素军负责拟定写作思路,指导撰写文章并最后定稿。
