金鱼源维氏气单胞菌JY01全基因组测序及生物信息分析
2021-09-22吴尉凤饶秋华柯文辉刘洋卓艺蓉郑欣欣罗土炎
吴尉凤 饶秋华 柯文辉 刘洋 卓艺蓉 郑欣欣 罗土炎
摘 要:維氏气单胞菌( Aeromonasveronii JY01)是从福州某养殖场内金鱼烂尾、腐皮和溃疡等病灶处分离出的1株革兰氏阴性优势菌。为了解其基因组结构及功能特征,基于IlluminaPE150和Pacbio测序平台对维氏气单肥菌菌株JY01进行全基因组测序,并对测序数据进行组装和组分分析,利用生物信息学手段进行基因预测与功能注释、同时预测致病因子和耐药基因。结果表明:维氏气单肥菌菌株JY01基因组序列长度为4.97 Mb,GC含量为58.17%;共预测到4601个编码基因、31个rRNA、16个基因岛及124个tRNA;其中,3461个基因在COG中获得注释,3082个基因聚集到GO聚类分析中,4345个基因在KEGG代谢通路中富集,预测维氏气单胞菌菌株JY01携带13种毒力因子包含240个相关毒力基因及9大类抗生素耐药性包含175个抗生素耐药基因。
关键词:金鱼;维氏气单胞菌;全基因组测序;基因注释
中图分类号:S 943 文献标志码:A 文章编号:0253-2301(2021)07-0018-08
DOI: 10.13651/j.cnki.fjnykj.2021.07.003
Whole Genome Sequencing and Bioinformatic Analysis of Aeromonas Veronii JY01 from Goldfish
WU Weifeng1, RAO Qiuhua2, KE Wenhui3, LIU Yang2, ZHUO Yirong1, ZHENG Xinxin1, LUO Tuyan2*
(1. Fujian Ocean Vocational College, Fuzhou, Fujian 350013, China; 2. Institute of Agricultural Quality Standards
and Testing Technology / Fujian Key Laboratory of Agroproducts Quality and Safety, Fujian Academy of
Agricultural Sciences, Fuzhou, Fujian 350003, China; 3. Institute of Agricultural Economics and
Scientific Information, Fujian Academy of Agricultural Sciences, Fuzhou, Fujian 350003, China)
Abstract: Aeromonas veronii JY01 is a strain of dominant gramnegative bacterium isolated from the lesions of goldfish, such as rotten tails, rot, and ulcers in a farm in Fuzhou. In this study, the whole genome of the strain of Aeromonas veronii JY01 was sequenced based on the IlluminaPE150 sequencing platform and Pacbio sequencing platform. The sequencing data was assembled and analyzed, the gene prediction and functional annotation were performed by bioinformatics methods, meanwhile the prediction of pathogenic genes and drug resistance genes were carried out. The results showed that the genomic sequence length of the strain of Aeromonas veronii JY01 was 4.97 Mb, and the GC content was 58.17%. A total of 4601 encoding genes, 31 rRNA, 16 gene islands and 124 tRNA were predicted, among which, 3461 genes were annotated in COG, 3082 genes were aggregated in GO cluster analysis, and 4345 genes were enriched in KEGG metabolic pathway. It was predicted that the strain of Aeromonas veronii JY01 carried 13 virulence factors including 240 related virulence genes, and 9 types of antibiotics resistance including 175 antibiotic resistance genes.
Key words: Goldfish; Aeromonas veronii ; Whole genome sequencing; Gene annotation
维氏气单胞菌 Aeromonas veronii 隶属于气单胞菌科Aeromonadaceae气单胞菌属 Aeromonas ,革兰氏染色阴性,端生或多生鞭毛短杆菌。该菌株由HiekmanBrenner等于1983年从临床腹泻病人的粪便中分离,于1987年利用DNAblot法首次鉴定为气单胞菌属内的新种[1]。因其能产生多种毒力因子,如气溶素、肠毒素以及菌毛、S层、荚膜、内毒素(LPS)和外膜蛋白(OMP)等粘附因子等[2-4],该菌株对人和多种水生动物具有很强的致病性。鱼类感染维氏气单胞菌后主要出现皮下点状溃烂、皮肤隆起、出血等症状。陆梦莹等[5]发现怀头鲶感染维氏气单胞菌后表现为明显出血、皮肤溃疡、鳍条充血、肛门红肿外突等临床症状;田甜等[6]发现中华鲟感染维氏气单胞菌后游动缓慢、食欲差且反应迟钝,体表口四周、眼眶、鳍条及侧骨板充血,肛门红肿,部分病鲟胸鳍出现蛀鳍现象。
本研究从福州某养殖场内金鱼烂尾、腐皮等病灶处分离纯化出1株维氏气单胞菌优势菌JY01。为了深入研究菌株JY01潜在的致病机制和耐药机制,用于流行病学、疫苗开发、微生物进化等领域。本研究采用第二代Illumina与第三代PacBio平台相结合的测序技术对维氏气单胞菌JY01进行全基因组测序,从而获得其全基因组序列,在KEGG、SwissProt、GO、NR和COG数据库中对菌株JY01基因功能进行注释、预测其致病因子和抗性基因,以期为金鱼维氏气单胞菌病的防控研究提供理论基础。
1 材料与方法
1.1 试验材料
1.1.1 试验菌株 维氏单胞菌菌株JY01来自实验室前期从自然发病的金鱼溃疡、烂尾和腐皮等病灶处分离的1株优势菌株,用于全基因组测序分析。
1.1.2 试剂材料 脑心浸出液培养基(BHI,BectoTM Brain Heart Infusion)购自BD中国代理机构,细菌基因组DNA提取试剂盒购自生工生物工程(上海)股份有限公司。
1.2 菌株的全基因组测序与组装
采用DNA提取试剂盒提取菌株JY01基因组DNA,其纯度和完整性利用琼脂糖凝胶电泳进行检测,采用Qubit方法进行定量。采用Covaris gTUBE将经电泳检测合格的DNA样品打断成构建文库所需大小的目的片段,按照PacBio SMRT全基因组DNA建库流程建立10 kb文库,建库后根据PacBio SMRT RS Ⅱ 测序平台上机流程进行全基因组测序;同时质量合格的DNA样品用Covaris超声波破碎仪随机打断成长度约为350 bp的片段,利用DNA文库构建(NEB, USA)试剂盒,构建350 bp小片段文库,步骤为:末端修复、加A尾、加测序接头、纯化、PCR扩增等,然后利用Illumina NovaSeq PE150测序。
过滤掉测序质量值低的reads,保留高质量reads即Clean Data。使用Unicycler软件(https://github.com/rrwick/Unicycler)[7]对Clean Data进行基因组组装,并将染色体序列组装成一个环状基因组完成图。
1.3 基因组组分分析
基因编码框采用GeneMarkS(Version 4.17)[8]软件从组装结果中进行预测,用于后续的基因功能注释分析。采用Tandem Repeats Finder(V.4.07b)软件[9]和RepeatMasker(V.4.0.5)
[10]软件分别对串联重复序列和转座子进行预测;通过tRNAscanSE软件(Version 1.3.1)[11]和rRNAmmer软件(Version 1.2)[12]分别预测tRNA和rRNA。采用CRISPRdigger(Version 1.0)[13]預测基因组中的CRISPR,使用IslandPathDIOMB 软件预测基因岛。
1.4 基因组的生物信息学分析
在已知的功能数据库中比对(evalue≤1×10-5)预测到的基因蛋白序列,在每条序列的比对结果中,选取score最高的比对结果(identity≥40%,coverage≥40%)作为基因功能注释的结果。参考数据库分别为非冗余的蛋白质数据库(Nr,NonRedundant Protein Database)[14]、SwissPro数据库[15]、基因本体论(GO,Gene Ontology)[16]、蛋白相邻类的聚簇(COG,Cluster of OrthologousGroups of proteins)[17]、碳水化合物活性酶数据库(CAZy,CarbohydrateActive enZymes Database)[18]和京都基因和基因组百科全书(KEGG,Kyoto Encyclopedia of Genes andGenomes)[19]功能数据库。
采用毒力因子数据库(VFDB,Virulence Factors of Pathogenic Bacteria)[20]和抗生素耐药基因数据库(CARD,the Comprehensive Antibiotic Research Database)[21]对菌株的基因组中所包含的毒力基因和耐药基因进行比对。
2 结果与分析
2.1 测序结果的统计及组装
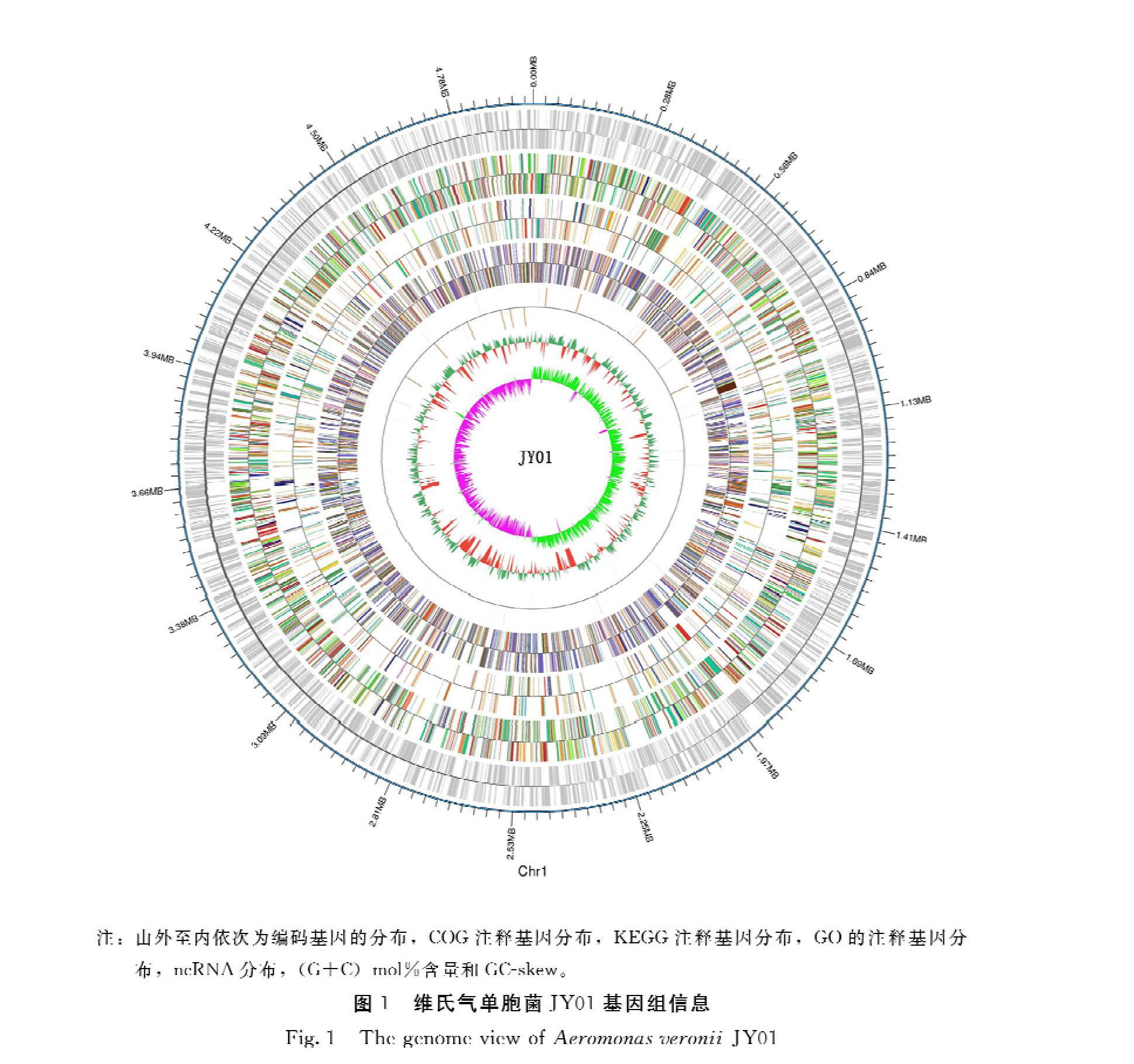
采用第二代Illumina平台与第三代PacBio平台相结合的测序技术,对菌株JY01全基因组进行测序。质控后共得到1 661 097 097 bp clean data,组装质控后的数据,并进行矫正和优化,最终获得基因组序列长度为4 969 544 bp,(G+C)mol%含量为58.17%,平均测序深度为334.26 X(图1)。
2.2 菌株JY01的基因结构
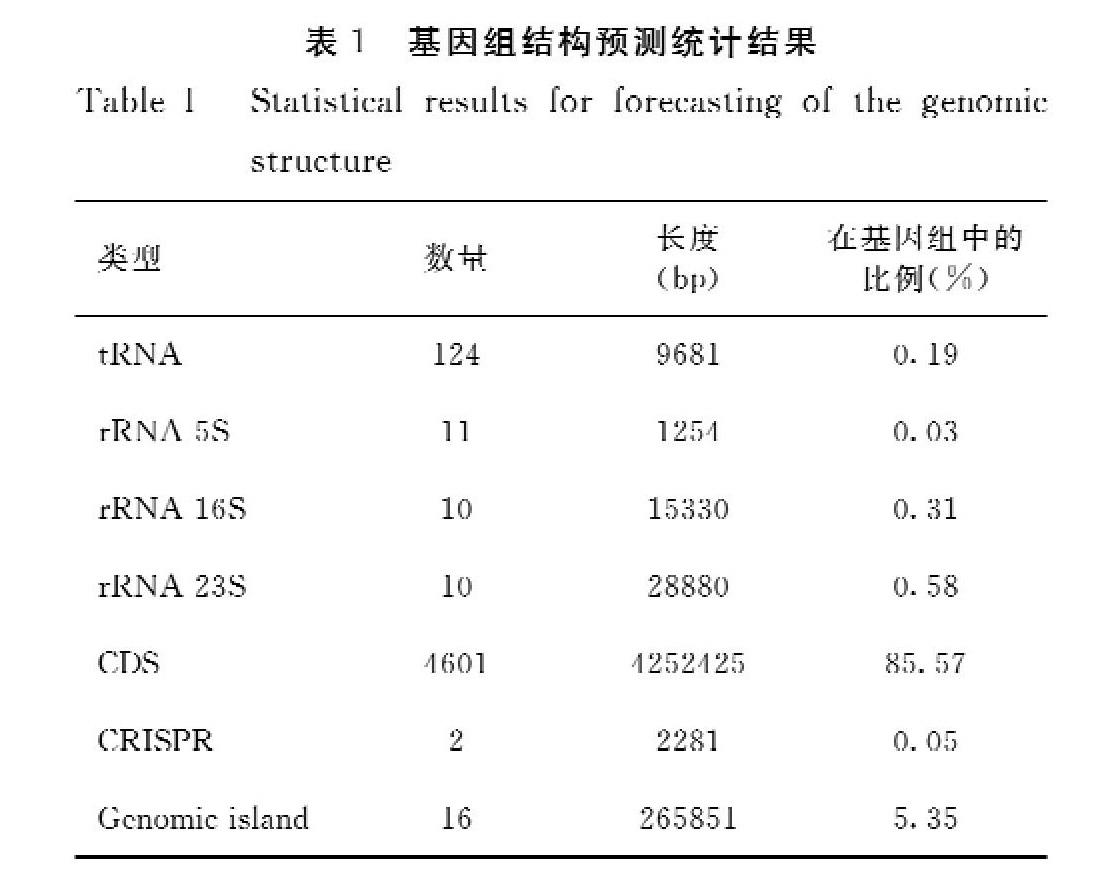
质粒是独立于核基因组可自主复制的环状DNA,普遍存在于原核生物细胞内。通过对组装后的测序结果进行分析,在JY01菌株未发现相应的质粒序列。原核生物中的CRISPR序列与Cas gene(CRISPR相关基因,CRISPRassociated genes)起到免疫系统(CRISPRCas系统)的作用,能识别并使入侵的功能元件沉默,经CRISPRdigger程序预测发现菌株JY01含有2组CRISPR相关序列。非编码RNA(ncRNA),如siRNA、snRNA、rRNA、tRNA等,从基因组上转录,可直接以RNA分子的形式发挥功能作用,菌株JY01中预测到的ncRNA 统计结果见表 1。基因岛编码蛋白具有多种功能,涉及发病机理和共生关系,还具有提高生物适应性等,菌株JY01基因组中预测到16个基因岛。
2.3 预测蛋白的数据库注释
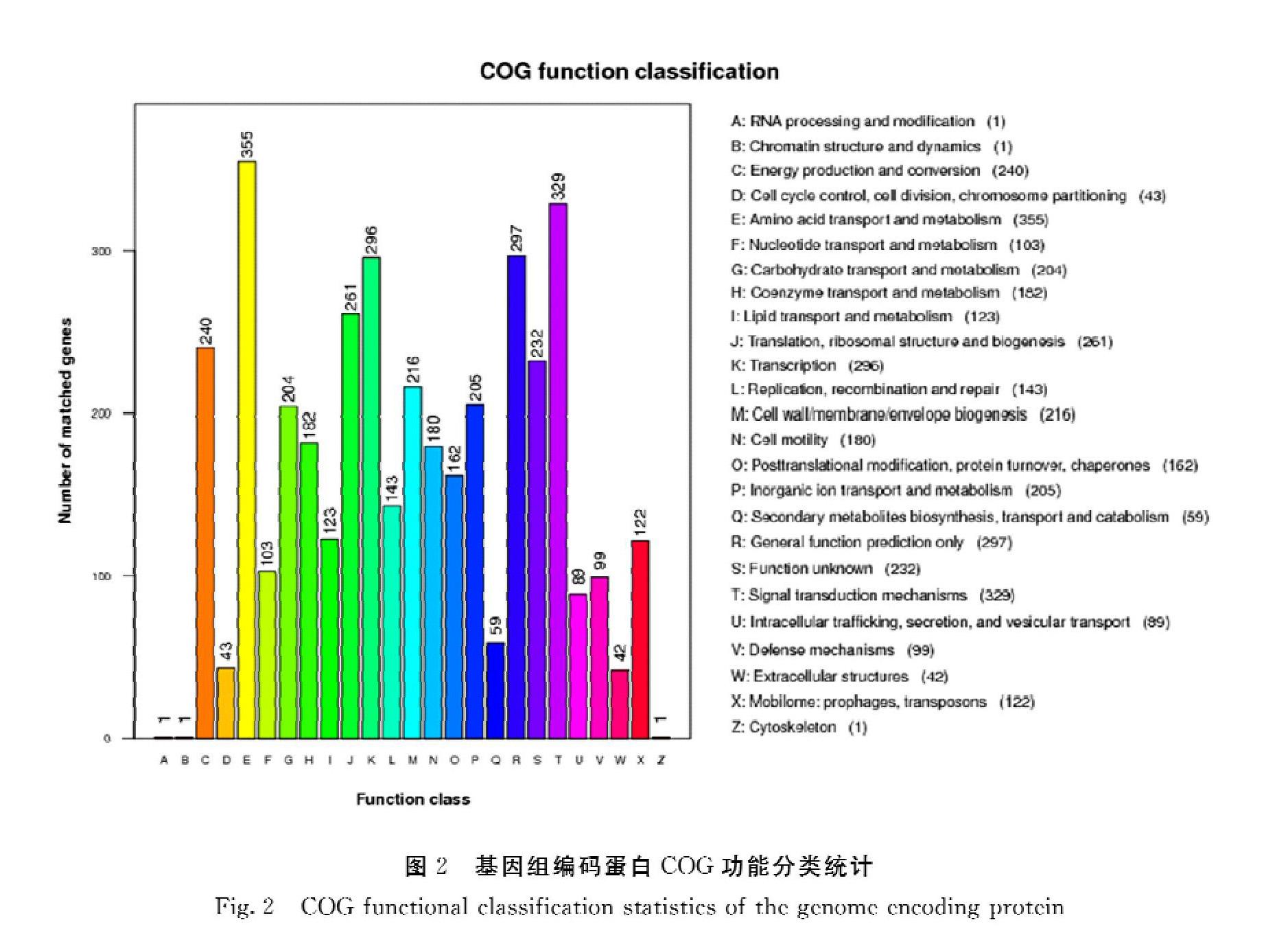
2.3.1 COG注释结果 维氏气单胞菌菌株JY01基因序列中预测到了4601个编码基因。在COG数据库得到注释的有3461个蛋白质,占总量的75.22%。菌株JY01中预测到的蛋白序列注释主要集中在以下功能(图2),分别为E(氨基酸的运输和代谢)355个,T(信号转导机制)329个,R(仅适用于一般功能预测)297个,K(转录)296个,J(翻译,核糖体结构与生物发生)261个,C(能源生产与转换)240个,S(未知功能)232个,M(细胞壁/膜/膜生物合成)216个,P(无机离子的转运与代谢)205个,G(碳水化合物的运输和代谢)204个,H(辅酶的运输和代谢)182个和N(细胞运动)180个。COG注释结果显示,关于碳水化合物的运输和代谢的基因达204个,另外有182个基因注释到辅酶运输代谢,表明该菌株可能具有糖类代谢及产多种辅酶的功能。
2.3.2 GO注释结果 维氏气单胞菌菌株JY01编码蛋白在GO数据库中得到注释共有3082个蛋白质序列,占总蛋白序列数量的66.98%。由图3可知,在分子功能方面,主要被注释到转运活性、结合、催化活性、活动分子传感器和核酸结合转录因子活性单元;在细胞组分方面,主要注释到细胞、细胞部分、细胞器、高分子配合物单元;在生物学过程方面,主要与细胞内过程、代谢过程、定位、定位建立、生物调节、刺激反应、生物过程调节、信号发射、细胞组成组织或生物发生单元有关。
2.3.3 KEGG注释结果 将测序结果与KEGG 数据库进行比对分析发现,菌株JY01预测到的基因有4345个富集到KEGG Pathway的217条代谢通路中,占基因总量的94.44%;其中含有大于50个基因数的代谢通路有15个(表2)。KEGG富集结果表明,代谢途径、次生代谢物的生物合成、抗生素的生物合成作为最主要的 3 条代谢通路,分别注释到620、285和194个基因;另外,微生物在不同环境中的代谢、双组分调节系统、碳代谢、ABC转运蛋白、氨基酸生物合成、嘌呤代谢通路与菌株JY01基因组具有较高的相关度。
2.3.4 CAZy功能分析 与CAZy数据库比对结果发现,菌株JY01的基因组中属于CAZy家族的共有151个基因编码的蛋白质结构域,其中包括40个蛋白基因属于13类糖苷转移酶家族(Glycosyl Transferases,GTs)、64个蛋白基因属于34类糖苷水解酶(Glycoside Hydrolases,GHs)、4个蛋白基因属于3类碳水化合物酯酶(Carbohydrate Esterases,CEs)、1个蛋白基因属于1类辅助酶类(Auxiliary Activities,AAs)、42个蛋白基因属于6类碳水化合物结合组件(CarbohydrateBinding Modules,CBMs)。菌株JY01基因组中未发现多糖裂解酶(Polysaccharide Lyases,PLs),可能与该菌株在淡水环境中的生境相关。
2.3.5 SwissProt数据库和NR数据库注释结果 菌株JY01的基因中序列与NR数据库进行alignment发现,在NR数据库中共有4431个基因得到相应注释,主要集中到 Aeromonas veronii、Aeromonas、Aeromonas hydrophila、Aeromonas allosaccharophila、Aeromonas jandaei、Aeromonas salmonicida、Aeromonas caviae和Edwardsiellaanguillarum 这8类物种,数量分别为2149、1760、149、68、46、28、27和20个。SwissPro是一个精选的蛋白质序列数据库,包括蛋白质结构、功能、翻译后修饰、变异等信息,菌株JY01中有2397个基因的蛋白序列功能在该数据库中得到有意义的注释。
2.3.6 毒力因子分析 与VFDB数据库alignment表明,菌株JY01共含有13种毒力因子及240个相关毒力基因,其中与细菌粘附功能相关的毒力因子有6个包含148个基因,说明该菌株对宿主具有较强的黏附能力,这是病原菌入侵的第一步;与分泌系统相关的毒力因子有3个包含88个基因;与毒素相关的毒力因子有4个包含4个基因(表3)。
2.3.7 耐药基因分析 CARD數据库注释结果发现,菌株JY01含有多种类型的抗生素耐药基因,包括9大类共175个抗生素耐药基因。从表4可知,外排泵系统基因数量最多,主要包括 macB (18个)、 patA (12个)和 lmrB (8个)等107个耐药基因。其次为多抗基因簇包含13个耐药基因,其余为介导磺胺类、多肽类抗生素(多粘菌素)、四环素类抗生素(四环素)、二氨基嘧啶类、β内酰胺类和喹诺酮类等抗生素耐药基因。
3 讨论
基因组测序伴随着生物信息学的发展,给研究微生物多样性、进化以及物种间相互作用等带来了全新的机会。在动植物病原菌的鉴定与分析方面,全基因组测序技术已广泛被引用,其可在分子水平上系统阐明致病菌的遗传进化地位、致病机理和与寄主的互作机制等,为病原菌的进化关系确定、耐药性的特征分析、预防策略的制定及疫苗开发奠定了理论基础。而维氏气单胞菌是一类兼性厌氧的革兰氏阴性杆菌,在水环境以及土壤中普遍存在,是一种条件致病菌,可同时够感染人类和鱼类,在水产业上常常给养殖业带来重大经济损失。因此,本研究旨在通过全基因组测序和生物信息学分析,了解金鱼源维氏气单胞菌JY01的基因组结构和功能,为进一步研究其致病机制和耐药机制提供重要生物信息学基础。
经denovo组装后,菌株JY01的基因组序列长度为4.97 Mb,共预测得到4601个编码基因,有4431个注释到已知功能蛋白,涉及转运、调控、环境适应和次级代谢活动等。GO注释结果表明,催化活性和结合等分子功能方面是菌株JY01的蛋白功能主要集中区,该功能方面的蛋白参与大量的代谢过程,同时包括对外界环境或自身的催化活性,为蛋白质的分泌过程和功能发挥提供了分子基础。COG分类具有局限性,菌株JY01内基因将近25%没有在COG里得到分类或功能注释,因此该基因组中有很多基因有待于更深层次的基因功能发掘。KEGG注释结果表明,菌株JY01的代谢途径丰富完整,能够使生物体自身及外界不断进行能量和物质交换,维持自身活动必需物质和对外界环境及时作出反应,增强病原菌的环境适应性。
细菌对抗抗生素机制主要包括:加快细菌主动外排作用,排出菌体内的抗生素;影响细胞膜渗透作用进而阻碍抗菌药物的进入;产生灭活酶或钝化酶破坏抗生素的活性;改变抗生素作用靶位等[22]。菌株JY01基因组上预测到9大类抗性基因,包含175个抗生素耐药基因,外排机制耐药基因数量最多,占61.14%。呈外排机制的耐药基因多数情况下可表现为多种抗菌药物的耐药性,如evgA、HNS、evgS同时具备RND和MFS两种耐药机制,TolC同时具备ABC、RND、MFS等3种耐药功能,soxS基因除兼备MFS3、RND、ABC等3种外排机制外,还具有降低抗生渗透性和改变抗生素作用靶点的功能。丰富的抗生素外排系统表明菌株JY01可能处于高浓度的抗生素环境中,巨大的选择压力导致其具有多种耐药性质。VFDB数据库比对发现,菌株JY01包含着多种毒力因子及相关毒力基因。主要集中在吸附和分泌系统上,分别占60.66%和36.07%。毒力因子中鞭毛、纤毛、粘附和侵袭、Ⅳ型菌毛提供运动占有重要的比例;而分泌系统能将蛋白整合在细胞膜或直接分泌到环境与靶细胞内,参与多种生理学过程,如病原菌的致病性、细胞黏附和环境的适应性等[23]。在毒力基因中,由某些基因共同调控形成的生物膜,构成了抗生素耐药的第一道防线机制。本研究对菌株JY01进行了全基因组测序与分析,有利于更全面地认识该菌株基因组的结构与功能,为该菌株的感染治疗和长期防控提供参考依据。
参考文献:
[1]HICKMANBRENNER F W, MACDONALD K L, STEIGERWALT A G,et al.Aeromonas veronii, a new ornithine decarboxylasepositive species that may causediarrhea[J]. J Clin Microbiol, 1987, 25(5): 900-906.
[2]MALTZ M, GRAF J.The type II secretion system is essential for erythrocyte lysis and gut colonization by the leech digestive tract symbiont Aeromonas veronii[J]. Appl Environ Microbiol, 2011, 77(2): 597-603.
[3]SEN K, LYE D.Importance of flagella and enterotoxins for Aeromonas virulence in a mouse model[J]. Can J Microbiol, 2007, 53(2): 261-269.
[4]MENCACCI A, CENCI E, MAZZOLLA R,et al.Aeromonas veronii biovar veroniisepticaemia and acute suppurative cholangitis in a patient with hepatitis B[J]. J Med Microbiol, 2003, 52(8): 727-730.
[5]陸梦莹, 胡秀彩.鲶源维氏气单胞菌的分离鉴定及药敏特性[J].大连海洋大学学报,2017,32(5):563-567.
[6]田甜, 张建明, 杜合军.中华鲟源维氏气单胞菌的分离鉴定及其药敏特性[J].安徽农业大学学报,2017,44(6):1010-1016.
[7]WICK R R, JUDD L M, GORRIE C L,et al.Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads[J]. PLoS Comput Biol, 2017, 13(6): e1005595.
[8]BESEMER J, LOMSADZE A, BORODOVSKY M.GeneMarkS: a selftraining method for prediction of gene starts in microbial genomes[J]. Nucleic Acids Res, 2001, 29(12): 2607-2618.
[9]BENSON G.Tandem repeats finder: a program to analyze DNA sequences[J]. Nucleic Acids Res, 1999, 27(2): 573-580.
[10]SAHA S, BRIDGES S, MAGBANUA Z V, et al.Empirical comparison of ab initio repeat finding programs[J]. Nucleic Acids Res, 2008, 36(7): 2284-2294.
[11]LOWE T M, EDDY S R.tRNAscanSE: a program for improved detection of transfer RNA genes in genomic sequence[J]. Nucleic Acids Res, 1997, 25(5): 955-964.
[12]LAGESEN K, HALLIN P, RODLAND E A, et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Res, 2007, 35(9): 3100-3108.
[13]GRISSA I, VERGNAUD G, POURCEL C.CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats[J]. Nucleic Acids Res , 2007, 35: 52-57.
[14]LI W,JAROSZEWSKI L, GODZIK A.Tolerating some redundancy significantly speeds up clustering of large protein databases[J]. Bioinformatics, 2002, 18(1): 77-82.
[15]BOECKMANN B, BAIROCH A, APWEILER R, et al.The SWISSPROT protein knowledgebase and its supplement TrEMBL in 2003[J]. Nucleic Acids Res, 2003, 31(1): 365-370.
[16]ASHBURNER M, BALL C A, BLAKE J A, et al.Gene ontology: tool for the unification of biology.The Gene Ontology Consortium[J]. Nat Genet, 2000, 25(1): 25-29.
[17]GALPERIN M Y, MAKAROVA K S, WOLF Y I, et al.Expanded microbial genome coverage and improved protein family annotation in the COG database[J]. Nucleic Acids Res, 2015, 43: 261-269.
[18]CANTAREL B L, COUTINHO P M, RANCUREL C, et al.The CarbohydrateActive EnZymes database (CAZy): an expert resource for Glycogenomics[J]. Nucleic Acids Res, 2009, 37: 233-238.
[19]KANEHISA M, GOTO S, KAWASHIMA S, et al.The KEGG resource for deciphering the genome[J]. Nucleic Acids Res, 2004, 32: 277-280.
[20]CHEN L, XIONG Z, SUN L, et al.VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors[J]. Nucleic Acids Res, 2012, 40: 641-645.
[21]JIA B F, RAPHENYA A R, ALCOCK B, et al.CARD 2017: expansion and modelcentric curation of the comprehensive antibiotic resistance database[J]. Nucleic Acids Res, 2017, 45(D1): D566-D573.
[22]史曉敏, 王少林.食品动物养殖环境中细菌耐药性研究进展[J].生物工程学报,2018,34(8):1234-1245.
[23]COSTA T R, FELISBERTORODRIGUES C, MEIR A, et al.Secretion systems in Gramnegative bacteria: structural and mechanistic insights[J]. Nat Rev Microbiol, 2015, 13(6): 343-359.
(责任编辑:林玲娜)
