天然混源萜Chromazonarol的合成及生物活性研究进展
2021-09-12李圣坤王侠
李圣坤 王侠

摘 要:Drimane 氢醌是来源于萜类和聚酮化合物途径的天然产物,它们通常具有稠环结构并显示多种重要的生物活性。Chromazonarol含有特征性的 6-6-6-6 四环骨架,且具有抗肿瘤、抗真菌、杀藻等重要生物活性,引起了众多有机合成化学家和药物化学家的关注。本文综述了天然产物Chromazonarol及异构体的分离,生物活性和合成进展。
关键词:Drimane 氢醌;生物活性;全合成
中图分类号:O629.6
文献标志码:A
天然产物具有特殊的结构特征和独特的生物活性机制,一直是发现和开发新医药[1]和农用化学品[2]的重要来源。Drimane氢醌类化合物,由补身烷骨架与苯基、取代苯基或氧化苯基(醌)组成,此类天然产物表现出抗肿瘤、抗菌、抗炎、抗HIV等重要的生物活性和药理活性,是新药创制的重要模型。天然产物Chromazonarol含有特征性的 6-6-6-6 稠合四环骨架,此类天然产物具有良好的药理活性,天然含量低且分离难度大,不能满足进一步药理学或者药物化学研究的需求。因此,通过来源易得的原料和有效的化学合成策略实现Chromazonarol类天然产物的制备,对实现此类天然产物的结构优化以及深入探讨构效关系和药理活性作用机制具有重要意义。鉴于此,本文介绍了Chromazonarol类天然产物的分离与生物活性,并对Chromazonarol类天然产物的合成进展进行了总结和对比。
1 天然混源萜Chromazonarol的分离和生物活性
Drimane混源萜类天然产物中很重要的一类是Drimane氢醌类化合物,其结构通式如图1-A所示(红色部分为drimane骨架、蓝色部分为苯基或醌),其中Chromazonarol含有特征性的 6-6-6-6 四环骨架(如图1-B所示),目前有3种立体异构体。1975年,CIMINO等[3]首次从海绵Disidea pallescens中分离出(+)-chromazonarol; 2012年, PRAWAT课题组从海绵Smenospongia sp中也分离出(+)-chromazonarol[4]。1975 年,FENICAL等[5]首次从海绵Disidea undulata中分离出(-)-chromazonarol;ISHIBASHI [6]和KUMAGAI [7]等課题组在对海绵Disidea undulata进行活性追踪的化学成分研究中也分离出(-)-chromazonarol。
1999年,BARRERO课题组首先测定并发现天然产物(+)-chromazonarol对肿瘤细胞P-388、A-549、HT-29、MEL-28具有不错的抑制活性,IC50值分别为15.91、15.91、15.91、15.91 μmol/L(如表1所示)。2004年,QUESADA等也测定了(+)-chromazonarol对内皮细胞和肿瘤细胞(BAEC、A-549、H116、PSN1、SKBR3、T98G)的生长影响,如表1所示。结果表明,(+)-chromazonarol显示出一定的抗肿瘤活性,并且对内皮细胞没有选择性[8-9]。
KUMAGAI等从海绵Disidea undulata1中分离出(-)-chromazonarol,并测定了它的1,1-二苯基-2-吡啶并肼基(DPPH)自由基清除活性。(-)-Chromazonarol显示出中等的DPPH自由基清除活性,EC50值为121 μmol/L[7]。ISHIBASHI等于2013年从海绵Disidea undulata中分离出(-)-chromazonarol,同时测定了它的杀藻活性,如表2所示。在1 μg/mL浓度处理下,(-)-chromazonarol对H. akashiwo和H. circularisquama显示出良好的杀藻活性,平均死亡率分别为78%和93%[6]。
此外,本课题组以廉价易得的二萜香紫苏醇,通过Barton脱羧偶联实现了(+)-chromazonarol的合成(见下节)并测试了它的抗真菌活性,发现(+)-chromazonarol对油菜菌核病菌(Sclerotinia scleotiorum)表现出良好的抗真菌活性,EC50值为24.1 μmol/L[10]。
研究表明,Chromazonarol类天然产物具有抗肿瘤、抗真菌、杀藻等重要生物活性。鉴于其重要的生物活性,这些天然产物自分离以来一直受到有机合成化学家的青睐。
2 Chromazonarol的合成进展
Chromazonarol类天然产物因其独特的结构和多种重要的药理活性引起了合成化学家和药物化学家的关注。目前针对Chromazonarol类天然产物发展的合成策略包括:芳基金属试剂对羰基的亲核加成、Friedel-Crafts烷基化、仿生环化、Diels-Alder环加成、Barton脱羧偶联以及 C(sp2)-C(sp3)偶联。
2.1 芳基金属试剂对羰基的亲核加成
BARRERO等[8]于1999年首次完成了(+)-chromazonarol的合成:以香紫苏醇4为起始原料,核心步骤是drimanic醛6与芳基锂化合物的亲核加成(图2)。首先香紫苏醇4经过四氧化锇-高碘酸钠氧化生成16个碳的乙酰氧基醛,再通过相应烯醇衍生物的氧化降解变成15个碳的醛中间体6,随后与化合物7的锂盐反应得到化合物8,用亚硫酰氯和吡啶进行脱水处理后产生区域异构体9 a-b (比例1∶ 2)。通过用氢氧化钾处理9 a-b的乙酸酯后,再用硼氢化钠进一步还原可得到10 a-b,接着用四丁基氟化铵处理,以高收率得到去保护的11 a-b,最后用三氟化硼乙醚环化便可非对映选择性得到(+)-chromazonarol。
2006年,VILLAMIZAR[11]等以(+)-manool为原料,通过相似的策略合成了(+)-chromazonarol(图3)。首先(+)-manool 12经过高锰酸钾氧化裂解成酮,在氧气存在下,再用叔丁醇钾氧化得到酸中间体13,经LiAlH4还原得到醇14,随后用四丙基高钌酸铵(TPAP)氧化得到醛15。利用间氯过氧苯甲酸处理化合物15,经Baeyer-Villiger氧化生成甲酸酯16,在氢氧化钾水解条件下得到(+)-albicanol 17,再用四丙基高钌酸铵氧化后便可得到关键中间体(+)-albicanal 18。在仲丁基锂条件下,18与芳基锂化合物发生亲核反应得到异构苄醇19。用Li/NH3/NH4Cl还原苄醇19得到化合物20,再经过脱保护,环化即可得到天然产物(+)-chromazonarol。
2013年,雷晓光[12]课题组利用亲核反应作为关键步骤也实现了(+)-chromazonarol的合成(图4)。与VILLAMIZAR方法相比,其是从香紫苏内酯制备,关键中间体醛22比以前使用(+)-manool作为起始原料的方法更有效。
2.2 Friedel-Crafts烷基化
2017年,DETHE小组[13]利用上文提到的相似的醛中间体,通过区域选择性脱水和还原获得关键的烯丙醇中间体23,通过三氟化硼乙醚(BF3·OEt2)催化的区域选择性傅克反应作为关键步骤方法合成了(+)-chromazonarol。如图5所示 [14],烯丙醇中间体23在Lewis酸条件下与富电子的二羟基苯甲醚24发生傅克反应,以良好的产率得到化合物25。在0 ℃下,用三氟甲磺酸酐和吡啶处理化合物25的酚羟基,得到三氟甲磺酸酯26。经过三氟甲磺酸酯基的还原裂解,得到甲基保护的(+)-chromazonarol 27,脱甲基后即可得到(+)-chromazonarol。
2.3 仿生环化
YAMAMOTO[15-17]课题组利用手性路易斯酸催化剂,通过立体选择性的多烯环化合成了天然产物(-)-chromazonarol(图6,图7)。利用此类催化剂((S)-LBA 3、(S)-3·SnCl4),可以实现4-苄氧基苯基法呢基醚28的串联环化,随后经Pd/C脱苄基保护,以40%的产率和44% ee得到(-)-chromazonarol。此路线较短,为制备苯并二氢吡喃骨架的多环萜类化合物提供了新思路。
针对上述路线中反应时间过长且选择性不高的问题, YAMAMOTO[18]课题组于2004年又设计了一种新的手性Lewis酸催化剂((S)-2e·SnCl4),利用其诱导化合物29选择性环化,最终以总收率39% 得到 (-)-chromazonarol(83% dr,91% ee )(图8)。较之前路线,选择性上有了很大提升。
2.4 Diels-Alder环加成
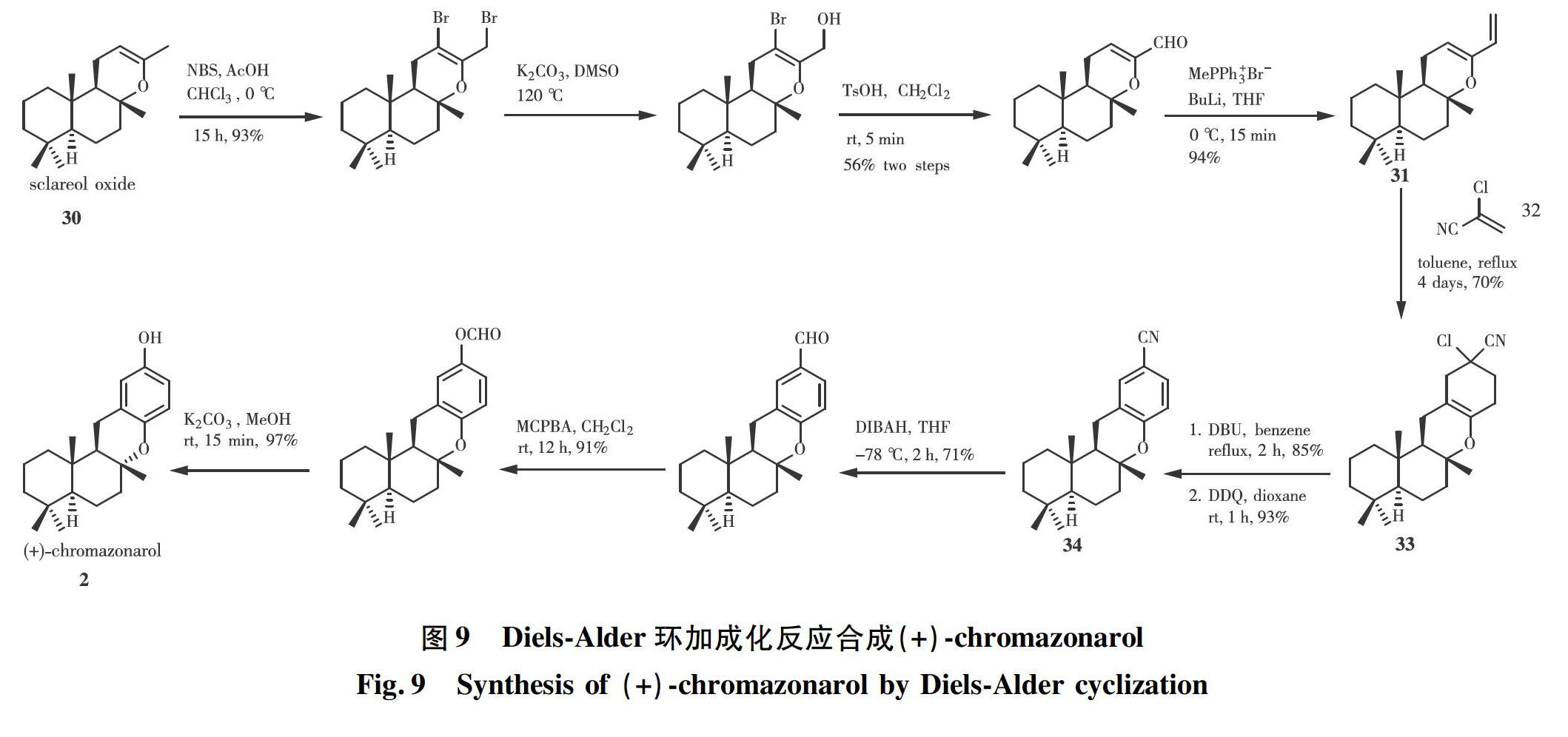
2007年,ALVAREZ-MANZANEDA[19]研究小组从香紫苏醇氧化物30出发合成了(+)-chromazonarol。如图9所示,以香紫苏醇氧化物30为起始原料,在溴代、碱性条件下,经过水解、氧化和Wittig反应等步骤合成了双烯体31,其在加热条件下与亲双烯体合成子氯丙烯腈32发生Diels-Alder环加成,实现6-6-6-6稠环化合物33的构建。依次经过1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)和二氯二氰苯醌(DDQ)处理,化合物33发生脱氯脱氢氧化得到芳香腈34,再经过还原、Baeyer-Villiger重排和水解等3步便可得到 (+)-chromazonarol。
2.5 Barton脱羧偶联
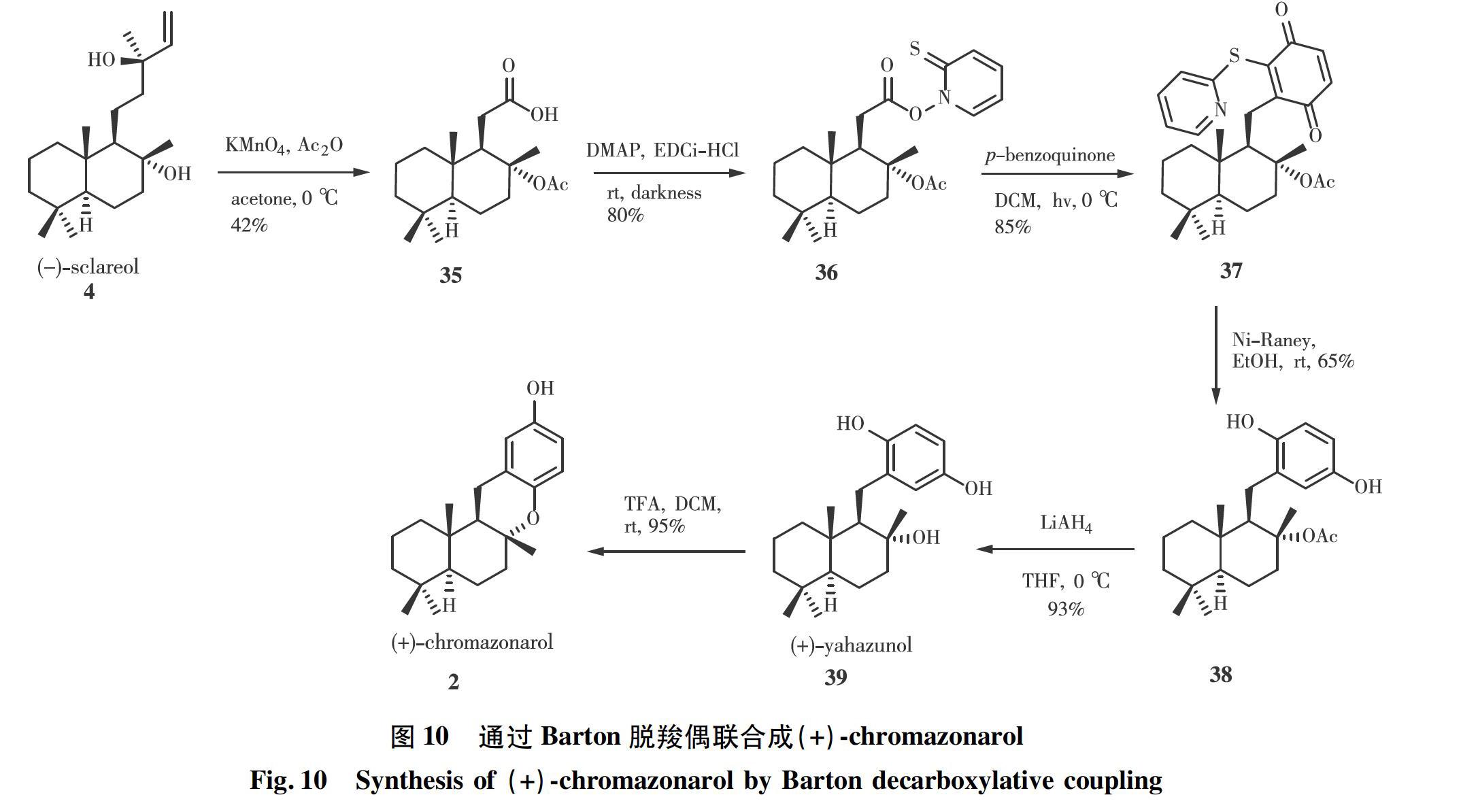
本课题组从香紫苏醇出发,以Barton脱羧偶联作为关键步骤合成了天然产物(+)-chromazonarol [10]。合成途径如图10所示。在醋酸酐(Ac2O)存在下,用高锰酸钾氧化降解香紫苏醇4,以中等收率得到酸35,酸35与2-巯基吡啶氮氧化物在Steglich酯化條件下缩合得到氧化还原活性酯36。活性酯与对苯醌在光照下发生脱羧偶联,生成化合物37,用雷尼镍处理便可得到乙酸酯保护的(+)-yahazunol 38,再用LiAlH4还原得到(+)-yahazunol 39。在室温下,用三氟乙酸(TFA)处理(+)-yahazunol,以94%的收率得到(+)-chromazonarol。该方法从香紫苏醇出发仅需3步即可构建连接Drimane和醌的共价键,且合成天然产物(+)-yahazunol仅需要5步。相比之下,通常将drimanyl醛与有机锂化合物偶联或在Suzuki偶联方法中将“硼杂香紫苏内酯”偶联需要6步以上。
2.6 C(sp2)-C(sp3)偶联反应模块化合成chromazonarol及类似物
2.6.1 通过“硼杂香紫苏内酯”合成 (+)-chromazonarol及类似物
2012年,BARAN[20]小组通过硼杂香紫苏内酯40合成了(+)-chromazonarol。如图11所示,化合物40从香紫苏内酯21出发,通过5步得到,其中包括:DIBAL-H介导的香紫苏内酯21的还原,PIDA/I2介导的C—C键氧化断裂、消除、水解和BH3硼氢化后,获得关键合成砌块硼杂香紫苏内酯40。在K2S2O8和AgNO3的条件下,通过用过量的1,4-苯醌处理40即可得到(+)-chromazonarol。
以硼杂香紫苏内酯40为中间体使得(+)-chromazonarol的合成比以前建立的方法更简洁,并可多样性地合成相关的Drimane氢醌类化合物。在合成硼杂香紫苏内酯40过程中,C8α-位置上的甲基会发生差向异构化,形成C8α-Me和C8β-Me差向异构体混合物。
2.6.2 鈀催化腙与芳基碘的交叉偶联
2018年,吴彦超[21]团队以香紫苏内酯21为原料,通过钯催化腙与芳基碘偶联的方法合成了天然产物(+)-chromazonarol。如图12所示,在-78 ℃下用六甲基二硅叠氮化钾(KHMDS)处理香紫苏内酯21,然后在P(OMe)3存在下与氧气反应,得到单一非对映异构体的α-羟基内酯。α-羟基内酯经过四氢铝锂、高碘酸钠处理后,再与对甲苯磺酰肼缩合得到所需的腙中间体41。通过钯催化的交叉偶联反应制备烯醇化合物42 a-b,用Et3SiH进行烯丙基还原和酸性条件下的脱水反应,以81%的产率得到化合物43。经过硝酸铈铵(CAN)氧化、连二亚硫酸钠还原得到化合物44,用三氟化硼乙醚(BF3·OEt2)进行环化即可得到(+)-chromazonarol。
2.6.3 Stille 羰基化偶联反应
2018年,厍学功团队[22]首次对映选择性全合成8-epi-chromazonarol。如图13所示,该路线以Stille羰基化交叉偶联反应作为核心步骤。由(R)-香芹酮45经过4步制备双环酮46[23],用KHMDS和苯基双(三氟甲烷磺酰)亚胺 (PhNTf2)处理得到双环三氟甲磺酸酯47。通过芳基锡烷48与双环三氟甲磺酸酯47的Stille 羰基化偶联反应实现drimane骨架与芳环的连接,完成了α,β-不饱和芳基酮49的合成。用三氟甲磺酸(TfOH)选择性脱苄基定量得到化合物50,由N2H4·H2O促进的分子内oxa-Michael环化反应实现chromazonarol的四环构建合成化合物51,再经过脱保护、脱羰后得到8-epi-chromazonarol。
2.6.4 脱羧硼化和Suzuki 偶联
本课题组开发了脱羧硼化与Suzuki 偶联相结合的一种策略,利用该方法可模块化地合成drimane氢醌类天然产物及其类似物。如图14所示,以香紫苏醇4为原料,经过三氯化钌和高碘酸钠氧化得到酸35,与N-羟基邻苯二甲酰亚胺缩合生成氧化还原活性酯52。在乙酰丙酮酸铜催化下,化合物52进行脱羧硼化得到关键合成子53,随后在Pd2(dba)3和RuPhos的催化组合下与溴代苯54进行Suzuki 偶联反应,以87%的产率得到苄基保护的(+)-yahazunol 55。根据之前报道的文献,化合物55再经过脱保护[24]、环化[10]2步便可得到(+)-chromazonarol。相比于BARAN小组的方法,该策略合成的关键中间体步骤短,中间体稳定且有良好的化学选择性。
3 总结与展望
本文对天然产物Chromazonarol及其异构体的分离和生物活性,该类天然产物的合成研究进展进行了综述。如表3所示,有机化学家从香紫苏醇、香紫苏内酯、泪杉醇等廉价易得的萜类原料出发,采用羰基与芳香金属试剂的亲核反应、Diels-Alder 环加成、Friede-Crafts 反应、C-C偶联以及仿生环化等关键步骤完成了Chromazonarol类天然产物的合成。
就目前所报道的全合成效率而论,其中羰基与芳香金属试剂的亲核反应、Diels-Alder 环加成、Friede-Crafts 反应合成路线多数较为冗长,有些转化需要采用贵金属试剂或催化剂、易燃试剂或超低温环境等不利于方便实用性制备目标分子的条件。仿生环化步骤虽短,但只能合成特定的几个天然产物,多样性不足;采用C(sp2)与C(sp3)偶联的策略则可高效模块化地合成Drimane氢醌类天然产物及相关类似物。本文总结了天然混源萜Chromazonarol的活性与合成研究进展,为结构相关Drimane氢醌类天然产物目标导向的合成、多样性制备和结构优化提供了很好的借鉴,为基于此类天然产物模型进行药物创制导向的化学生物学以及药理学研究打下很好的基础。
参考文献:
[1]NEWMAN D J, CRAGG G M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019[J]. Journal of Natural Products, 2020, 83(3): 770-803.
[2]SPARKS T C, HAHN D R, GARIZI N V. Natural products, their derivatives, mimics and synthetic equivalents: role in agrochemical discovery[J]. Pest Management Science, 2017, 73(4): 700-715.
[3]CIMINO G, DE STEFANO S, MINALE L. ent-Chromazonarol, a chroman-sesquiterpenoid from the sponge Disidea pallescens[J]. Experientia, 1975, 31(10): 1117-1118.
[4]PRAWAT H, MAHIDOL C, KAWEETRIPOB W, et al. Iodo-sesquiterpene hydroquinone and brominated indole alkaloids from the Thai sponge Smenospongia sp[J]. Tetrahedron, 2012, 68(34): 6881-6886.
[5]FENICAL W, MCCONNELL O. Chromazonarol and isochromazonarol, new chromanols from the brown seaweed Dictyopteris undulata (zonarioides)[J]. Experientia, 1975, 31: 1004-1005.
[6]ISHIBASHI F, SATO S, SAKAI K, et al. Algicidal sesquiterpene hydroquinones from the brown alga Dictyopteris undulata[J]. Bioscience, Biotechnology, and Biochemistry, 2013, 77(5): 1120-1122.
[7]KUMAGAI M, NISHIKAWA K, MATSUURA H, et al. Antioxidants from the Brown Alga Dictyopteris undulata[J]. Molecules, 2018, 23(5): 1214.
[8]BARRERO A F, ALVAREZ-MANZANEDA E J, HERRADOR M M, et al. Synthesis and antitumoral activities of marine ent-chromazonarol and related compounds[J]. Bioorganic & Medicinal Chemistry Letters, 1999, 9(16): 2325-2328.
[9]CASTRO M E, GONZáLEZ-IRIARTE M, BARRERO A F, et al. Study of puupehenone and related compounds as inhibitors of angiogenesis[J]. International Journal of Cancer, 2004, 110(1): 31-38.
[10]ZHANG S S, WANG X, HAO J, et al. Expediently scalable synthesis and antifungal exploration of (+)-yahazunol and related meroterpenoids[J]. Journal of Natural Products, 2018, 81(9): 2010-2017.
[11]VILLAMIZAR J, PLATA F, CANUDAS N, et al. New access to sesquiterpene hydroquinones: synthesis of (+)-ent-chromazonarol[J]. Synthetic Communications, 2006, 36(3): 311-320.
[12]HUANG J H, LEI X G. A nature-inspired concise synthesis of (+)-ent-chromazonarol[J]. Science China Chemistry, 2013, 56(3): 349-353.
[13]DETHE D H, MURHADE G M, DHERANGE B D, et al. Enantiospecific syntheses of hongoquercins A and B and chromazonarol[J]. European Journal of Organic Chemistry, 2017, 2017(7): 1143-1150.
[14]GEORGE J H, BALDWIN J E, ADLINGTON R M. Enantiospecific, biosynthetically inspired formal total synthesis of (+)-liphagal[J]. Organic Letters, 2010, 12(10): 2394-2397.
[15]ISHIBASHI H, ISHIHARA K, YAMAMOTO H. Chiral proton donor reagents: tin tetrachloride: coordinated optically active binaphthol derivatives[J]. Chemical record (New York, NY), 2002, 2(3): 177-188.
[16]NAKAMURA S, ISHIHARA K, YAMAMOTO H. Enantioselective biomimetic cyclization of isoprenoids using Lewis acid-assisted chiral bronsted acids: abnormal claisen rearrangements and successive cyclizations[J]. Journal of the American Chemical Society, 2000, 122(34): 8131-8140.
[17]ISHIHARA K, NAKAMURA S, YAMAMOTO H. The first enantioselective biomimetic cyclization of polyprenoids[J]. Journal of the American Chemical Society, 1999, 121(20): 4906-4907.
[18]ISHIBASHI H, ISHIHARA K, YAMAMOTO H. A new artificial cyclase for polyprenoids: enantioselective total synthesis of (-)-chromazonarol, (+)-8-epi-puupehedione, and (-)-11-deoxytaondiol methyl ether[J]. Journal of the American Chemical Society, 2004, 126(36): 11122-11123.
[19]ALVAREZ-MANZANEDA E J, CHAHBOUN R, CABRERA E, et al. Diels-alder cycloaddition approach to puupehenone-related metabolites: synthesis of the potent angiogenesis inhibitor 8-epipuupehedione[J]. The Journal of Organic Chemistry, 2007, 72(9): 3332-3339.
[20]DIXON D D, LOCKNER J W, ZHOU Q, et al. Scalable, divergent synthesis of meroterpenoids via “Borono-sclareolide”[J]. Journal of the American Chemical Society, 2012, 134: 8432-8435.
[21]WANG H S, LI H J, ZHANG Z G, et al. Divergent synthesis of bioactive marine meroterpenoids by palladium-catalyzed tandem carbene migratory insertion[J]. European Journal of Organic Chemistry, 2018, 2018(7): 915-925.
[22]LIU L, SONG H Y, CHEN P, et al. Total synthesis of (-)-8-epi-chromazonarol enabled by a unique N2H4·H2O promoted intramolecular oxa-Michael cyclization reaction[J]. Organic Chemistry Frontiers, 2018, 5(20): 3013-3017.
[23]GESSON J P, JACQUESY J C, RENOUX B. A new chiral route toward terpenoids. Annulation of carvone to trans- and cis-fused bicyclic synthons[J]. Tetrahedron, 1989, 45(18): 5853-5866.
[24]LAUBE T, BEIL W, SEIFERT K. Total synthesis of two 12-nordrimanes and the pharmacological active sesquiterpene hydroquinone yahazunol[J]. Tetrahedron, 2005, 61(5): 1141-1148.
(責任编辑:周晓南)
Abstract:
Drimane quinone/hydroquinone sesquiterpenes are natural products of mixed biosynthetic origin which are derived from both terpenoid and polyketide pathways. They usually possess fused polycyclic systems and display multiple interesting biological activities. Chromazonarol features a characteristic 6-6-6-6 tetracyclic framework, exhibiting antitumor, algicidal and antifungal activities. Chromazonarol has attracted a great deal of attentions from both synthetic organic chemists and medicinal chemists. This review summarizes the isolation, biological activities and synthesis of chromazonarol.
Key words:
Drimane quinone/hydroquinone sesquiterpenes; biological activities; total synthesis
李圣坤,男,1984年生,博士,教授,博士生导师,一直从事新农药导向的原创性配体发掘及化学生物学研究。基于病原微生物呼吸电子传递链,以芳基噁唑啉和Drimane混源萜类天然产物为模型,提出了去芳香化和仿生化策略,实现了原创性药物先导的发掘,构思了多功能配体骨架设计理念,完成了重要药物先导的精准制备,并在上述基础上实现了成果转化或技术合作。
