12例海外华人SARS-CoV-2感染者病毒基因组测序及变异分析
2021-09-09陈建威崔恩海胡国海王晓光季巧英杨伟斌
曾 永,陈建威,崔恩海,胡国海,王晓光,季巧英,丛 迎,杨 阳,杨伟斌
(1. 青田县疾控中心,青田 323900;2. 青岛华大基因研究院,青岛 266555;3. 湖州市中心医院,湖州 313000;4. 深圳国家基因库,深圳 518083;5. 丽水市疾控中心,丽水 323000;6. 丽水市中心医院,丽水 323000)
2019年12 月,湖北省武汉市出现严重急性呼吸综合征冠状病毒2(severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)引起的新型冠状病毒肺炎(corona virus disease 2019,COVID-19)[1-2]。病毒感染随后蔓延到全国各地,导致严重的呼吸道疾病,严重危害人们的生命健康[3]。除中国外,COVID-19疫情在全球多个国家也出现了,成为国际关注的突发公共卫生事件[4-5]。同时,中国开始出现海外输入性病例,防控病毒的战斗还在继续。
SARS-CoV-2在人群中的持续传播可能会产生适应性突变,因此极有可能会累计基因突变,进而抵抗人类的免疫系统[6]。已有研究表明,基因突变可能会导致新型冠状病毒的毒力、传染性和传播性的改变[7-8]。因此,对不同来源的新冠肺炎病例进行基因组分析有助于了解SARS-CoV-2的变异与感染病程的相关性,从而为疾病预防和控制提供建议。
鉴于患者咽拭子和痰液中RNA含量的差异,我们采用最新研发的捕获测序[9-10]和宏转录组测序方法[10],对意大利归国和国内的新冠肺炎确诊病例进行基因组突变分析。与宏转录组测序相比,捕获测序所需数据量仅为宏转录组测序的1/10或更少,且测序结果准确。对病毒基因组的突变分析为深入研究意大利归国患者SARS-CoV-2的致病力、传染性以及国内毒株的差异打下了基础。
1 材料与方法
1.1 患者标本收集、RNA提取和实时逆转录聚合酶链反应(qRT-PCR)定量
本项目涉及的所有临床标本(包括咽拭子和痰液)均来自浙江省新冠肺炎确诊的19例患者,其中7例为国内病例,12例为欧洲输入性病例(11例为意大利输入性病例,1例为西班牙输入性病例)。在11名意大利患者中有7名是意大利贝加莫同一家餐厅的工作人员(详细临床资料见表1)。该项伦理审查已获丽水市中心医院和深圳华大生命科学研究院伦理委员会(institutional review board,IRB)批准。所有参与者在纳入这项研究之前都签署了书面知情同意书。
通过Maxwell™16 Total RNA纯化试剂盒进行SARS-CoV-2 RNA提取。随后针对SARS-CoV-2的RdRp基因和N基因进行实时逆转录聚合酶链反应(quantitative real-time PCR,qRT-PCR)。采用HCoV-19核酸检测试剂盒检测和定量检测临床标本中的病毒RNA的载量(捷诺生物,上海)。
1.2 宏转录组文库的制备和测序
宏转录组测序技术可及时快速地对传染病进行检测、防控和变异分析[11-12]。首先,我们对每个RNA样本用DNase I去除DNA,并对纯化的产物进行定量(Qubit RNA HS Assay Kit, Thermo Fisher Science, Waltham, MA, USA);然后将纯化的RNA在87℃缓冲液中孵育6 min进行打断,随后将RNA片段利用随机引物合成双链(ds)cDNA并进行末端修复、接头连接;最后对(ds)cDNA进行18个循环的PCR扩增并加上双端测序标签(unique dual-indexed adapters,UDI),使用MGIEasy RNA(华大智造,深圳)的试剂盒经过环化、滚环复制后构建DNA纳米球(DNA nanoball,DNB)文库[10]。使用Qubit 3.0 荧光定量仪 (Invitrogen, Carlsbad, CA, USA)检测文库浓度,并使用Agilent 2100 生物分析仪检测文库片段大小,对合格文库采用DNBSEQ T1测序平台(华大智造,深圳)或MGISEQ-2000测序平台(华大智造,深圳)进行双端100 bp 读长模式的高通量测序[4]。
1.3 病毒杂交捕获富集和测序
病毒全基因组杂交捕获技术是病毒分离鉴定的常用方法之一,可提升检测灵敏度和病毒基因组覆盖度[13-14]。利用MGIEasy外显子捕获辅助试剂盒(华大智造,深圳)替代了SARS-CoV-2 DNA/RNA捕获试剂盒(伯科生物,北京)中的阻滞剂寡核苷酸(blocker oligos)和PCR引物寡核苷酸(PCR primer oligos)后,采用基于混合捕获的富集方法从宏转录组逆转录双链cDNA片段中富集SARS-CoV-2特异性片段,并根据试剂盒说明对SARS-CoV-2病毒全基因组进行扩增[10]。给富集扩增得到的病毒cDNA片段加上双端测序标签,经过环化、滚环复制后构建DNB文库,对每个库检合格的文库使用MGISEQ-2000测序平台(华大智造,深圳)进行双端100 bp 读长模式的高通量测序[4]。
1.4 SARS-CoV-2病毒序列鉴定
为了获得高质量的SARS-CoV-2病毒测序序列,我们采取了4个严格的质量控制步骤,从每个宏转录组文库和杂交捕获文库测序结果中获得高质量的SARS-CoV-2片段序列。首先用Kraken(v0.10.5)软件[15]将原始测序数据同SARS-CoV-2参考基因组(GISAID获取号:EPI_ISL_402119)[16]数据进行对比,筛选得到候选的SARS-CoV-2原始测序数据;然后依次用fastp(v0.19.5,参数:-q20-u20-n1-150)、SOAPnuke(v1.5.6,参数:-l20-q0.2-E50-d)和PRINSEQ(v0.20.4,参数:LC_Method Dust-LC_Threshold 7)对测序数据的低质量数据、接头污染、PCR重复测序序列和低复杂度序列进行过滤。如果该文库最终高质量有效测序序列数小于1 000条,则舍弃其测序数据,不用于下游分析。针对每个样本,本文将两个不同建库方法获得的大规模并行高通量测序(massively parallel sequencing,MPS)数据合并用于变异检测、组装和其他下游分析。使用Samtools (v1.9)计算SARS-CoV-2参考基因组在所有文库测序数据中每个核苷酸位点的覆盖深度。对每个文库的测序数据以200 bp的滑动窗口用R(v3.6.1)软件中的ggplot2包绘制SARS-CoV-2参考基因组的测序覆盖率和测序深度。
1.5 病毒基因组组装及单核苷酸变异(SNVs)检测
对于参考覆盖深度大于15 X(X表示全基因组的测序覆盖深度的层数)的样本,截取最多100 X的SARS-CoV-2测序数据,使用SPAdes(v3.14.0)软件[17]默认参数进行组装得到完整的病毒基因序列。对于其他低深度样本,使用IDBA_hyrid(v1.1.3,参数为“-mink 21--maxk 91--step 10”)进行病毒基因组组装[18]。
为了检测每个病毒样品的单核苷酸变异(single nucleotide variations,SNVs)信息,首先使用BWA aln(v0.7.16)将每个文库的有效测序数据与SARSCoV2参考基因组(EPI_ISL_402119)进行比对,然后使用Picard (v2.10.10)去除重复的比对结果。基于去重的比对结果使用FreeBayes (v1.3.1,参数:-p 1-q 20-m 60-V)[19]分析得到每个测序数据的SNVs,最后使用snippy-vcf_filter(参数:--minqual 100--minfrac 0.8;https://github.com/tseemann/snippy)过滤低置信度SNVs。剩余的高置信度SNVs在SARS-CoV-2参考基因组中用SNVeff(v4.3)[20]的默认参数进行注释。如果测序数据的参考基因组覆盖深度大于15 X,则所有SNVs变异检测调用软件的最小覆盖参数设置为10 X;如果测序覆盖深度小于15 X,则设置为4 X。为了评估每个样本的SNVs准确性,使用pysamstats (v1.1.2)软件(https://github. com/alimanfoo/pysamstats)和Pilon(v1.23)软件[21]进行变异检测,并保留至少两种方法检测到的SNVs。对于高深度测序(>40 X)的样本,对BWA软件比对结果使用与Ni等[22]相同方法及参数进行宿主内单核苷酸变异(intradividual single nucleotide variations,iSNVs)检测。
2 结果与分析
2.1 病毒基因组测序
我们通过qRT-PCR检测19个SARS-CoV-2样本的Ct值,结果表明所有样本的Ct值均在22.43~38.84之间(表1)。其中仅3例样本(C06、F03和F12)检测到高病毒拷贝(Ct<24.5),6例样本(C05、F05、F04、C01、C07和C02)检测到低病毒拷贝(24.5<Ct<28.7),其余10例样本检测到极低病毒拷贝(Ct>28.7)。

表1 19个新冠病例样本临床信息Tab. 1 Detailed clinical information of 19 cases
对所有SARS-CoV-2 RNA样本构建逆转录文库,并对其全基因组进行了测序。这些样品获得的原始测序数据双端序列条数在8 995万条到140 824万条之间,平均约为64 968万条(130 Gb)。用Kraken和BWA对SARS-CoV-2病毒测序数据进行分类,并计算每个文库数据的覆盖深度。虽然每个文库均产出了大量的测序数据,但由于病毒拷贝数较低,在Ct<28.7的6个样本中,仅有0.004 5%~0.055 8%的测序数据被归类为SARSCoV-2病毒序列(比例最低的样本C01和最高的样本C05除外),而在Ct>28.7的11个样本中,仅有0.001 4%~0.006 2%的测序数据被归类为SARSCoV-2。在19个测序结果中,只有10个样本的有效参考基因组覆盖率超过95%,且测序覆盖深度大于5 X(表2)。
然后,我们构建了14个基于杂交捕获的文库并进行测序以获得病毒的基因组数据,其中包括9个宏转录测序数据基因组覆盖度不足的样本(F05、F10、F01、C04、F02、F06、C03、F09和F11)和5个宏转录测序数据基因组高覆盖度的样本(C06、F12、C05、F04和F07,包含不同的Ct值)。杂交捕获测序的每个样本平均得到了约7 000万条病毒测序序列条数,分类鉴定得到SARS-CoV-2病毒测序数据的平均比率为8.4%(表2),其比例显著高于宏转录组测序所得的结果,表明捕获测序法更适合于高Ct(低病毒载量)样本的测序。该方法测序的所有样品覆盖深度均大于5 X,基因组覆盖率为95%(表2),可满足用于突变和组装分析。
2.2 宏转录组测序和杂交捕获测序的SNVs检测比较
为了比较两种不同建库方法大规模并行MPS结果的准确性,我们使用宏转录组和杂交捕获方法分别对5个样本(C06、F12、C05、F04和F07)进行了文库构建及测序。分析表明,每个文库测序数据的参考基因组深度都超过了5 X,且参考基因组覆盖率均大于96%(图1a、表2)。使用Pilon和FreeBayes方法对每个测序数据进行SNVs检测,取两个方法都认为是可靠的SNVs作为该测序数据检测到的SNVs。结果表明,不论在高病毒拷贝数样本(C06、F12)中,还是在低病毒拷贝数样本(C05、F04)和极低病毒拷贝数样本(F07)中,两个不同建库方法测序数据检测到的SNVs完全一样,具有很高的一致性(表3)。上述结果显示,相对直接进行宏转录组测序,杂交捕获测序对不同Ct值的样本均具有准确的突变检测结果,且所需样本量仅为宏转录组测序的1/10或更少。高Ct(低病毒载量)的样品即使测序超过100 Gb也不能获得有效的SARS-CoV-2病毒测序信息,但捕获测序可获得有效的信息,表明杂交捕获测序是一种更适合于低病毒载量样本的检测方法。

表2 19个新冠病例样本宏转录组测序数据和杂交捕获测序数据与参考基因组比对统计Tab. 2 Sequencing data and SARS-CoV-2 reference genome mapping statistics

表3 宏转录组测序和杂交捕获测序检测的SNVs变异信息Tab. 3 SNVs detection by metatranscriptomic and hybrid capture
2.3 基因组组装及SNVs分析
合并两个不同建库方式的测序数据后,总共有10个样品的测序深度大于15 X,其余9个样品测序深度为5~15 X,全部样品的参考基因组覆盖率均大于93%(图1b)。使用SPAdes或IDBA_hyrid进行组装,共得到10个完整的SARS-CoV-2病毒基因组序列和9个SARS-CoV-2草图基因组序列,其中9个草图基因组序列含有12~2 174 bp的gaps(表4)。除F09的覆盖率为93%外,所有组装的基因组相对于参考基因组的覆盖率均在95%以上。
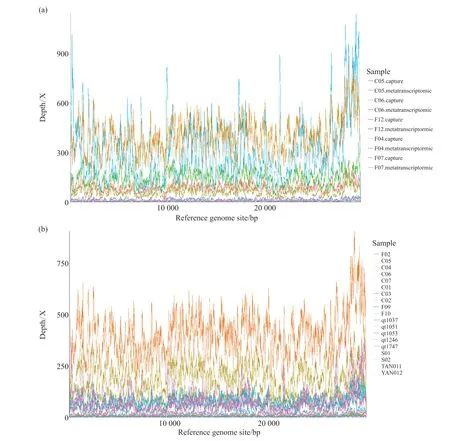
图1 SARS-CoV-2参考基因组(EPI_ISL_402119)覆盖深度情况Fig. 1 The sequencing coverage depth of SARS-CoV-2 reference genome (EPI_ISL_402119)(a)5个不同Ct样本宏转录组测序和杂交捕获测序参考基因组覆盖深度;(b)19个样品全部测序数据统计参考基因组覆盖深度。(a) The sequencing depth of 5 different Ct samples by metatranscriptomic sequencing and hybrid capture sequencing; (b) Statistical sequencing depth for all sequencing data of 19 samples.
使用FreeBayes、Pysamstats和Pilon 3种方法对19个样本进行SNVs检测,取至少2种方法检测到的高置信SNVs为最终鉴定的SNVs (表5)。在19个样品中总共检测到了ORF1a:C409S、ORF1a:P1207H、ORF8:L84S和S:D614G 4种突变型,分布在基因ORF1a、ORF8和S上(表4、5)。突变分支S(Clade S,ORF8:L84S)包括4个中国样本(C01、C02、C07和C03)和唯一的西班牙样本F01,这些样品都是在基因ORF8的第251位碱基由T变成C,导致病毒第84位编码氨基酸由亮氨酸(Leu,L)突变为丝氨酸(Ser,S)。突变分支G(Clade G,S:D614G)包括来自同一家意大利餐厅的4个样本(F07、F08、F09和F10)以及另外5个意大利样本(F05、F02、F03、F04和F12)。这9个意大利样本的S蛋白具有相同的突变,都是SRAS-CoV-2病毒第614位氨基酸由天冬氨酸(Asp,D)变成了甘氨酸(Gly,G)。此外,具有相同突变ORF1a:P1207H的样本包括2个来自中国庆元市的样本(C05和C06)和2个来自同一家意大利餐厅的样本(F06和F12),具有突变ORF1a:C409S的只有1个来自中国的样本C04。变异分析表明,在中国及意大利的新型冠状病毒肺炎患者检测到的SARS-CoV-2病毒基因组均存在不一样的变异类群。同时样本间检测具有差异的SNVs,表明了SARS-CoV-2病毒变异的多态性。

表4 病毒基因组组装和突变类型统计Tab. 4 Genome assembly and mutation clade statistics

表5 (续)Tab. 5 (continued)
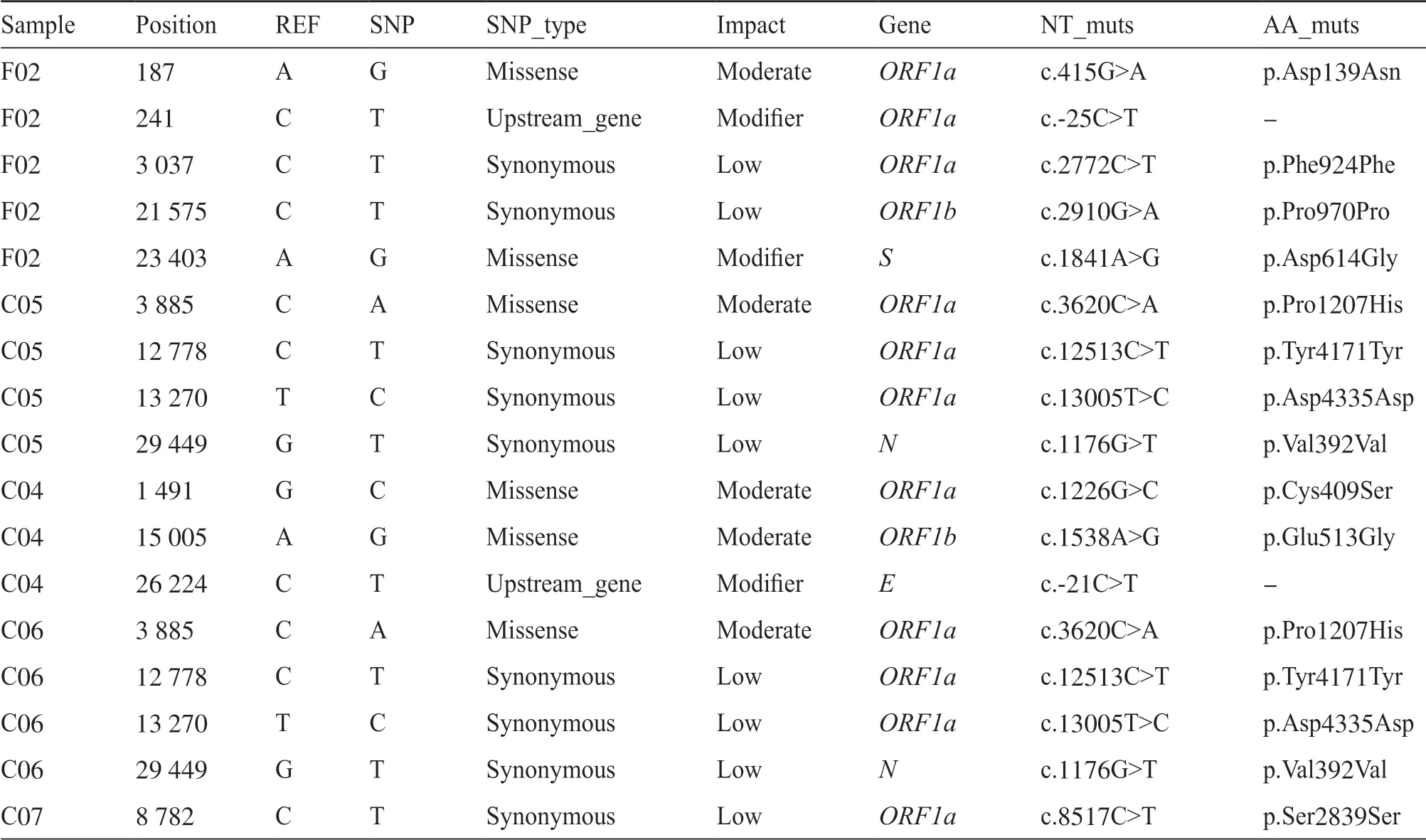
表5 19个新冠病例样本SNVs变异检测结果Tab. 5 SNVs detection of 19 sequencing SARS-CoV-2 samples

表5 (续)Tab. 5 (continued)
2.4 iSNVs变异检测
我们对高测序深度(>40 X)的9个样本进行了iSNVs分析。结果表明:在F03、F08和C06 3个样本中没有检测到任何iSNVs;在C02、C07、C05和F04这4个样本中至少检测到一个iSNVs,但没有iSNVs位于基因区或非翻译区(untranslated region,UTR)。此外,在F06和F11这2个样本中均检测到了4个iSNVs,都具有相同的突变c.1841A>G(p.Asp614Gly,即S:D614G)(表6)。F06和F11样本基因区的iSNVs与来自同一家意大利餐厅的其他5个病例样本的SNVs突变结果意外一致,表明在同一患者中可能存在重复感染。
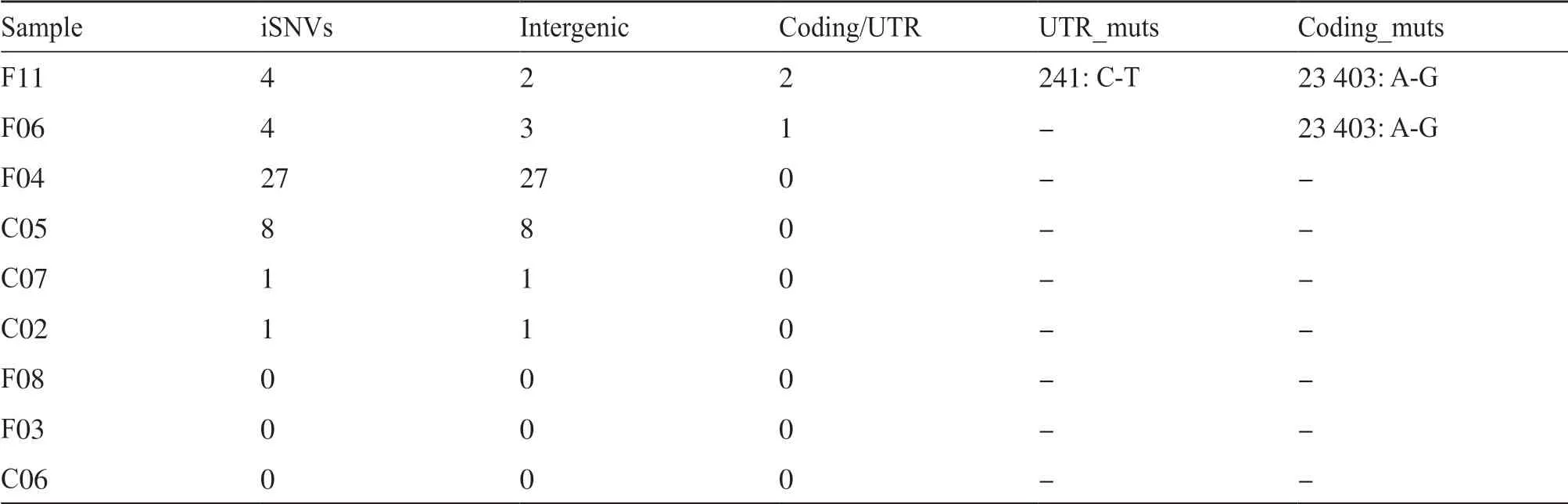
表6 高深度测序样品iSNVs变异信息统计Tab. 6 The statistics of iSNVs for high depth sequencing samples
3 讨论
本研究从新冠肺炎病例咽拭子和痰液样本中提取Total RNA,并采用宏转录组和杂交捕获方法进行高通量全基因组测序,最终获得了19个样本有效的高通量测序数据,组装得到了10个完整的新冠病毒基因组序列和9个新冠病毒基因组草图。通过比较宏转录组测序和捕获测序的参考基因组覆盖度以及变异检测情况,证实了捕获测序对于不同病毒载量的样品均可获得有效、准确的组装和变异检测结果。
此外,通过变异结果分析,11个意大利归国华人中9个病例均检测到了S蛋白D614G突变,且具有D614G突变的样本大部分都伴有5'UTR中的C到T突变(相对于参考基因组EPI_ISL_402119的241位)、3 037位的C到T突变、14 408位的C到T突变。另外2个病例虽然直接检测到的变异型不属于D614G,但在iSNVs检测中也均发现了D614G突变。具有D614G突变的SARS-CoV-2病毒株2020年2月初开始在欧洲传播[23]。我们3月初采集的来自意大利不同地区的F02、F03、F04、F07、F08、F10和F12 7个样品都能检测到同时含有这4种突变的相同突变型(表5),表明在3月初之前具有这4个遗传连锁突变的单倍型已在意大利不同地区开始传播流行。根据目前研究,D614G突变毒株已成为世界范围内的主流毒株,在欧洲和北美的测序样本中占近70%,且其具有更强的感染性和传播性[24],这可能是意大利疫情暴发的一个重要因素。
我们对19个样本进行SNVs检测,结果共发现4种突变,其中意大利患者中分离的病毒株是具有高传染性的S:D614G突变病毒株,而ORF1a:C409S、ORF1a:P1207H和ORF8:L84S这3个突变主要在中国人群中被检测到,可见SARS-CoV-2病毒突变在意大利和中国并不相同。鉴于SARS-CoV-2基因组会不断的发生突变,那么对SARS-CoV-2基因组的测序就显得极为重要,而我们新研发的捕获测序的方法与传统的宏转录组测序相比所需的样本量更少,是一种更适合于低病毒载量样本的检测方法。通过捕获测序获得SARS-CoV-2病毒的突变信息并研究突变的功能,对于后续抗体和疫苗设计具有重要的指导意义。
