亚麻全基因组关联分析的研究进展
2021-09-02陆美光段海燕姜恭好
陆美光,段海燕,姜恭好
(1黑龙江大学现代农业与生态环境学院,哈尔滨 150080;2黑龙江大学生命科学学院,哈尔滨 150080)
0 引言
亚麻(Linum usitatissimumL.)是亚麻科亚麻属一年生或多年生的双子叶自花授粉的草本植物,根据用途可分为油用型亚麻、纤维用型亚麻和两用型亚麻,中国西北地区和华北地区都将亚麻视作重要的油料和经济作物[1]。亚麻是五大油料作物之一,且作为当今世界第三大纤维作物[2],在经济作物生产中,亚麻具有十分重要的地位,中国的纤维亚麻收获面积在世界占据第一位[3]。亚麻的栽培历史十分悠久,其起源于中东、地中海,亚麻的原产地之一也包括中国[4],因其适应性很强,所以在世界各地都有分布[5],亚麻广泛生长在加拿大、美国、印度、德国以及俄罗斯和中国,纤维亚麻主要生长在北欧、俄罗斯和中国[6]。在中国,甘肃、内蒙古和新疆是亚麻的主要种植地,黑龙江省亚麻总产量占全国的85%[7]。亚麻栽培已有五千多年的历史[8],是人类最早使用的植物天然纤维及油料作物[9]。几千年来,亚麻一直被人类用作高品质油和茎纤维的来源[10]。
亚麻用途广泛,功能多样,近年来在纺织、工业、医药保健、食品、新材料等很多方面具有重要用途。亚麻生长的条件加上一些调节剂的使用,能够促进亚麻对重金属的吸收;由于其纤维强度高,吸湿散热能力好,并且具有防静电的特性,又将其用于复合材料、汽车内装饰等[11]。亚麻籽中亚麻籽油、亚麻籽蛋白、亚麻籽胶、木脂素等营养成分能够有效调节脂质代谢、降低血糖血脂水平、预防癌症[12],在药品、食品、保健品和化妆品中被广泛应用[13]。此外,亚麻秆中含丰富的矿物元素,其中富含钾、梓等人体必需的微量元素。亚麻饼粕粗蛋白质和总磷含量高,并且总磷真利用率较高,是较好的蛋白质和磷源饲料原料[14]。
全基因组关联分析(genome-wide association study,GWAS)是应用基因组中数以百万计的SNP为分子遗传标记,进行全基因组水平上的对照分析或相关性分析,并通过比较发现影响复杂性状的基因变异的新技术,从而在分子水平揭示作物表型多样性的遗传基础,为作物性状的改良提供理论基础,为分子育种提供新的途径[15]。GWAS在亚麻多种性状研究以及育种中得到了一定的应用,但相较于水稻、玉米等作物研究较少,本文综述了国内外亚麻GWAS的研究进展,并简要综述了其他作物GWAS研究进展,以期为亚麻GWAS研究提供理论依据及新思路。
1 全基因组关联分析
1.1 全基因组关联分析优势
关联分析(Association Analysis),又称关联作图(Association mapping,AM),是以连锁不平衡(linkage disequilibrium,LD)为基础,将群体内的遗传标记与目标性状的表型数据进行关联的分析方法,用来发掘与目标性状相关联的遗传位点[16]。连锁不平衡是指分属两个或两个以上基因座位的等位基因同时出现在一条染色体上的频率高于随机出现的频率。只要两个基因不是完全独立地遗传,就会表现出某种程度的连锁,这种情况就叫连锁不平衡。连锁不平衡可以是同一条染色体上的不同区域,也可以是不同染色体上的区域[17]。GWAS具有三点主要优势[18]:(1)检测范围广,同时检测多个性状或同一座位的多个等位基因。GWAS是对全基因组范围内的所有位点进行关联分析,因此其拥有更广泛的关联信息;(2)精确度高,分辨率高。自然群体经过多次重组后,连锁不平衡衰减,增加了定位的精确性,提高了基因鉴定的效率;(3)群体构建时间短。GWAS通常选择自然群体作为材料,相对传统QTL定位要求的遗传群体所需的构建时间更短。随着高通量测序成本的降低,目前全基因组关联分析已经成为研究作物复杂性状的有效手段之一。
1.2 全基因组关联分析流程
全基因组关联分析流程见图1。
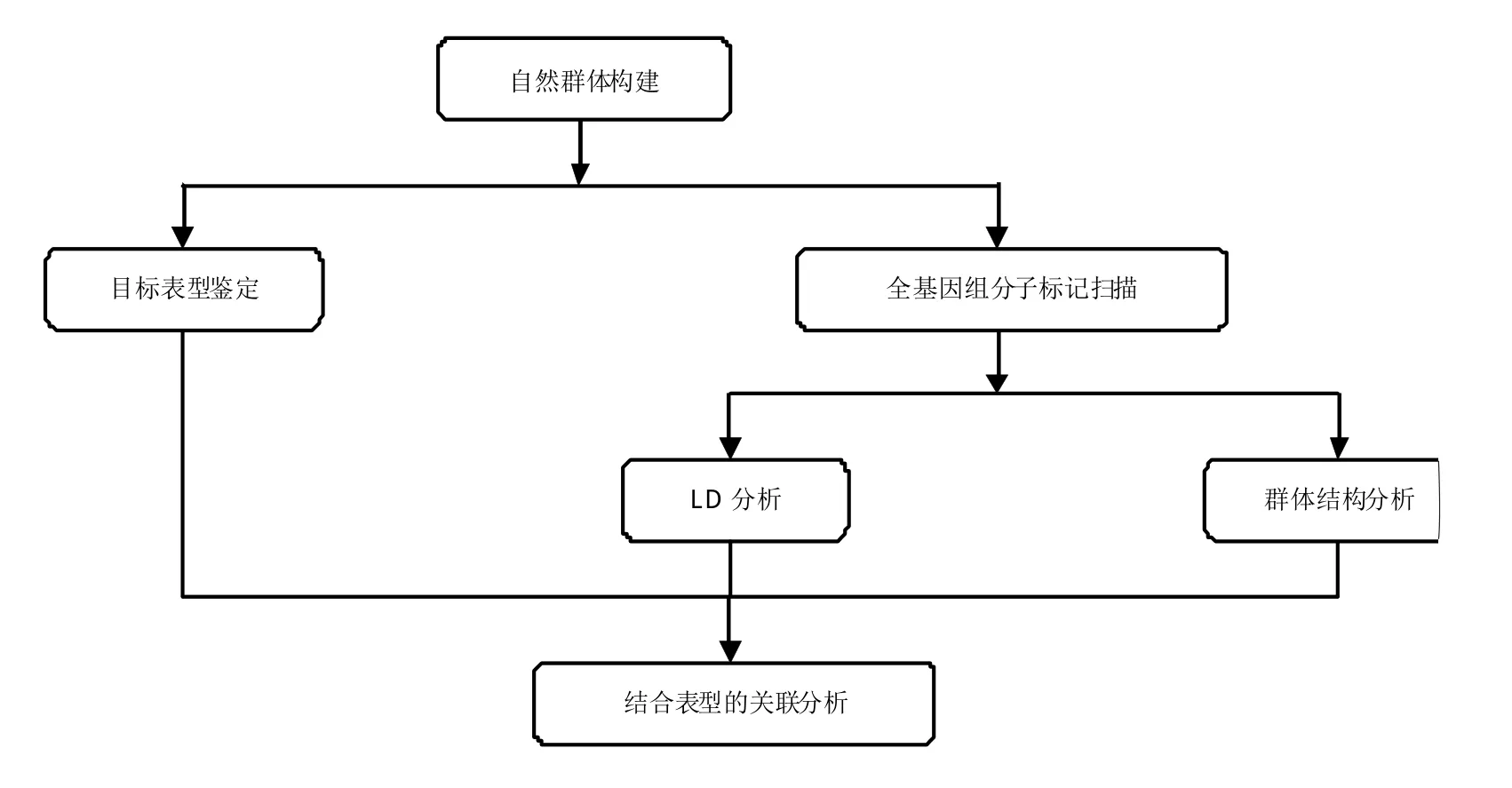
图1 GWAS技术路线图
(1)收集遗传种质材料,构建自然群体[19]。具有丰富遗传变异的遗传群体是关联分析的遗传基础,因此自然群体要尽可能多地来自不同地区。自然群体还要有足够大的样本量,尽可能包括该物种的全部遗传变异信息,自然群体样品数目一般要大于200个。
(2)目标性状表型鉴定[20]。表型性状不仅受基因型的控制,也会受到周围环境的影响,因此对GWAS的分析结果产生很大的影响,因此,为了尽可能减少误差,目标性状的表型鉴定要在多年和多环境中进行。
(3)基因型分析。随着测序技术的快速发展,高通量测序成本降低,全基因组范围内分子标记的密度目前多采用覆盖全基因组的高密度的单核苷酸多态性(SNP)标记,可极大地提升关联分析遗传定位的精度。
(4)关联分析。利用遗传模型对目标性状的表型数据和基因型数据进行关联分析,最终检测控制目标性状遗传特性的遗传位点。
2 GWAS在亚麻中的应用
2.1 产量相关性状
亚麻的产量相关性状主要包括株高、千粒重、工艺长度、分枝数等。产量相关性状能够决定亚麻的产量,因此,研究产量相关性状的遗传机理能够有效地提高亚麻的产量,缓解中国亚麻的市场需求。目前,国内外已有研究者在GWAS基础上对亚麻的产量相关性状进行研究。邓欣[4]利用SSR标记对182份亚麻核心种质进行关联分析,共检测到10个关联位点。其中包括与株高关联的1个位点,与工艺长度关联的1个位点,与分枝数关联的2个位点,与单株果数关联的1个位点。伊六喜[21]对401份胡麻(油用亚麻)种质进行表型变异和SRAP标记的遗传多样性分析,以此为基础构建了关联群体,并通过重测序技术对269份胡麻关联群体进行全基因组关联分析,最终获得了产量相关性状的显著SNP位点21个,候选基因57个;其中株高相关SNP有5个,并获得了14个候选基因;工艺长度4个SNP并获得了15个候选基因;分枝数2个SNP并获得了5个候选基因;株果数1个SNP并获得了7个候选基因;千粒重3个SNP并获得了5个候选基因;果粒数2个SNP并获得了8个候选基因;单株粒重2个SNP并获得了1个候选基因;全生育日数2个SNP并获得了2个候选基因;其中显著性SNP对表型的解释率为6.76%~12.97%。Soto-Cerda[22]等对390份加拿大核心种质亚麻材料的464个SSR标记进行基因型分析,以及来自8个环境收集的包括株高、千粒重、分枝数等9个农艺性状的表型数据进行关联作图分析,共鉴定出6个农艺性状的12个性状关联的显著标记位点,且定位到1个与抗倒伏相关的QTL。Xie等[23]首先测量了224份核心亚麻种质3种环境下的株高、工艺长度、分枝数、单株果数和千粒重的表型数据,随后又以其作为关联群体利用SLAF-seq对这5个农艺性状进行全基因组关联分析,共检测出9个与株高显著相关的SNP位点,3个与工艺长度显著相关的SNP位点,21个与分枝数显著相关的SNP位点,9个与单株果数显著相关的SNP位点,23个与千粒重显著相关的SNP位点,并利用GLM和MLM模型获得15个候选基因,最终确定UGT和PL是株高的候选基因,GRAS和XTH是分枝数的候选基因,Contig1437和LU0019C12是单株果数的候选基因,PHO1是千粒重的候选基因。随后又对13个农艺性状进行了表型分析[24],再通过全基因组关联作图来分析和鉴定这些性状的潜在遗传位点,EMMAX模型和GLM模型被用于不同的性状关联研究,最终检测到单株果数数、千粒重、棕榈酸、硬脂酸、亚油酸、亚麻酸6个性状的16个SNP位点,并推测Lus10016125是亚麻株高的候选基因。
2.2 品质相关性状
亚麻能够为人类提供亚油酸、亚麻酸、木酚素等物质,可被应用于医疗保健领域,因此,对亚麻品质相关性状进行遗传学研究有助于提高亚麻品质,是进行亚麻优质育种的基础。伊六喜[21]对401份胡麻(油用亚麻)种质进行表型变异和SRAP标记的遗传多样性分析,以此为基础构建了关联群体,并通过重测序技术对269份胡麻关联群体进行全基因组关联分析,最终获得了19个品质相关性状相关的显著SNP位点以及43个候选基因;其中包括8个亚麻酸SNP并获得17个候选基因;2个粗脂肪SNP并获得了3个候选基因;3个硬脂酸SNP并获得了7个候选基因;3个油酸SNP并获得了5个候选基因;1个亚油酸SNP并获得了6个候选基因;2个棕榈酸SNP并获得了5个候选基因;其中显著SNP的表型解释率为5.62%~11.04%。张喻[25]利用200份来自43个国家的优质亚麻品种作为关联群体,对脂肪酸的组成与含量进行统计分析,并通过全基因组测序获得的多个SNP位点进行GWAS,发掘亚麻籽中所有与脂肪酸合成以及代谢相关的候选基因,最终共发掘到了8个与油酸代谢明显相关的候选基因以及2个与亚油酸代谢相关的候选基因。Soto-Cerda等[26]利用460个SSR标记对390份亚麻核心种质进行基因型分析,又从390份种质材料中收集表型数据,分别用2个一般线性模型和3个混合线性模型对含油率、棕榈酸、硬脂酸、油酸、亚油酸、亚麻酸和碘值7个品质性状进行关联作图分析,最终定位到5个相关性状的9个QTL,其中包括4个高度稳定的QTL,棕榈酸和油酸没有检测到QTL,但LG2上的标记lu2046和LG6上的标记lu2555分别解释了8.4%和3.9%的变异。Xie等[24]对224份亚麻核心种质的13个农艺性状进行GWAS,利用EMMAX模型和GLM检测到单株果数数、千粒重、棕榈酸、硬脂酸、亚油酸、亚麻酸6个性状的16个SNP位点,并确定Lus10022606为棕榈酸的候选基因,验证了Lus10017450可能是控制亚油酸合成的候选基因。随后,Xie等[27]基于SLAF-seq对种子脂肪酸含量进行全基因组关联分析,共检测到16个与种子脂肪酸含量显著相关的SNP位点,并筛选出10个候选基因,其中6个候选基因参与了重要的脂肪酸代谢途径且与5种脂肪酸的含量显著相关。You等[28]对260份亚麻品系进行GWAS,共鉴定了17288个单核苷酸多态性,解释了80%以上的成熟期、碘值、棕榈酸、硬脂酸、亚油酸和亚麻酸含量的表型变异,并利用GLM和MLM模型共鉴定出包括粗脂肪、碘值、棕榈酸、亚油酸、亚麻酸等11个性状相关的33个QTL,解释了48%~73%的表型变异。
2.3 其他性状
除了产量相关性状和品质相关性状,GWAS还在亚麻一些其他性状上得到了应用,见表1。He等[29]为了确定亚麻派斯莫病抗性相关的遗传区域,对370份亚麻核心种质进行了全基因组关联研究,鉴定出分布在亚麻15条染色体上的258873个单核苷酸多态性,并利用10种统计模型最终检测到67个相关QTL,解释了32%~64%的变异率。Soto-Cerda等[30]利用200份亚麻种质材料作为关联群体对种子粘液含量和籽外壳含量进行全基因组关联分析,最终获得12个种子粘液相关的SNP和17个籽外壳含量相关的SNP,并定位到7个和4个分别与两种性状相关的QTL。Chandrawati等[31]利用SSR标记对168份亚麻种质材料进行群体结构和全基因组关联分析,并利用GLM和MLM模型关联分析出Lu_3043是与50%开花天数相关的标记。伊六喜等[32]为挖掘胡麻木酚素含量的相关基因,利用269份胡麻种质作为关联群体,对其基因型数据和木酚素含量数据进行全基因组关联分析,最终共获得13个显著SNP位点和21个候选基因,此研究为胡麻分子标记辅助育种和高木酚素含量新品种选育提供了科学依据。

表1 亚麻各性状GWAS相关进展
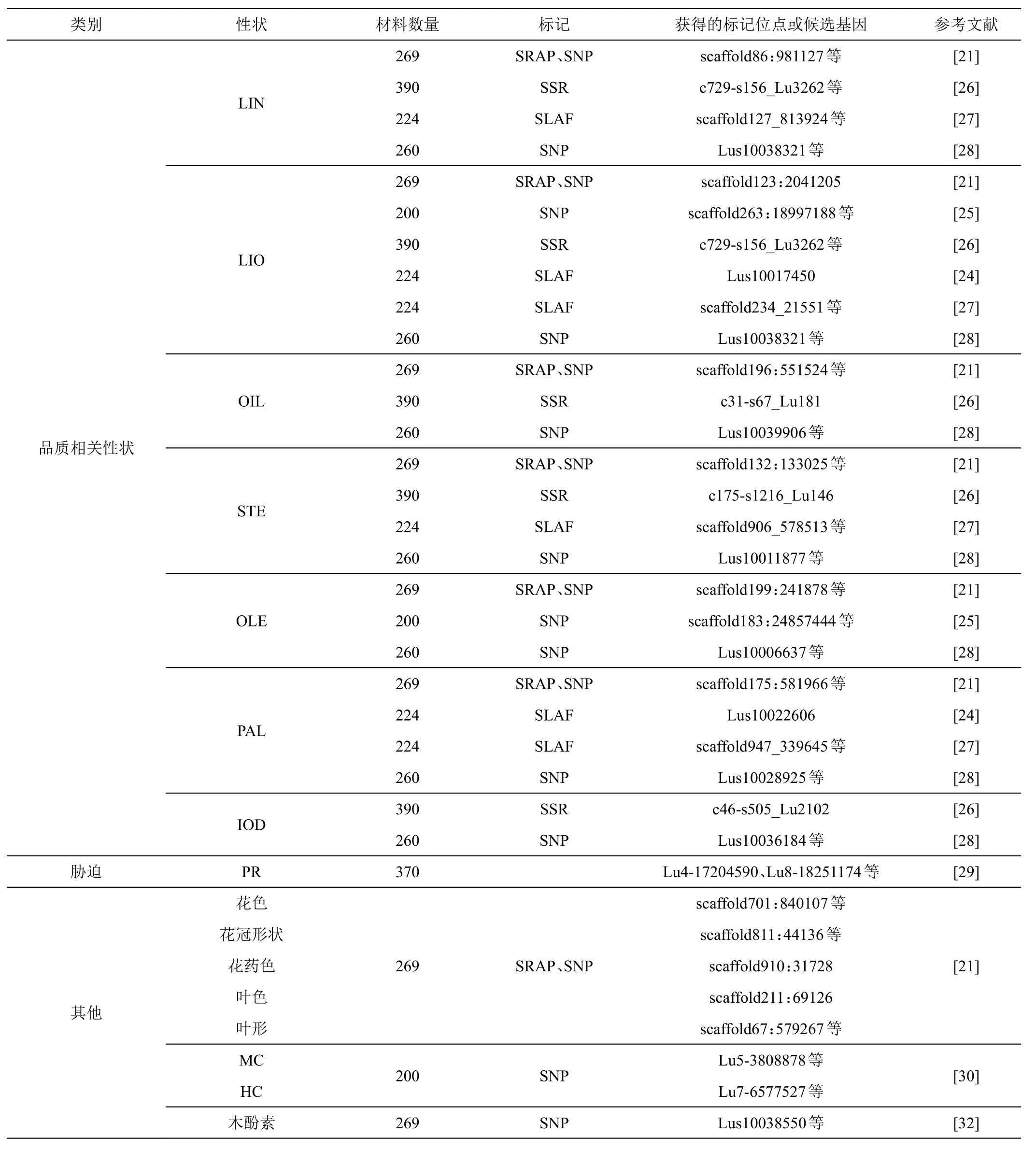
续表1
2.4 亚麻白粉病抗性GWAS研究
亚麻GWAS遗传学研究起步较晚,但随着亚麻基因组研究的不断发展和关联分析方法的不断进步,GWAS技术将会被更多地应用于亚麻重要性状的遗传学研究。亚麻主要病害有白粉病、锈病、派斯莫病等十余种,这些病害导致亚麻生产面积逐渐降低,品质下降,单产不高,总产不稳,且这些病害还有逐年加重的趋势[33],研究并探索与病害相关的抗性基因有助于缓解病害带来的减产压力。白粉病是亚麻的一种常发病,在自然条件下全田发病仅需20天,其发病时间短、流行速度快。病菌首先发生在叶片正面,再扩大到叶片背面及叶柄处,最后扩散到全叶,病叶会出现提前变黄,卷曲枯死的现象,严重影响的亚麻产量和质量[33]。到目前为止,已经有研究者对亚麻抗白粉病的遗传机制进行研究,已发现的亚麻白粉病的抗性大部分均由1个显性基因控制,其中被命名的基因有3个[34],分别为 pm1[35]、ol[36]、pm-linum[37]。Asgarinia[38]等,利用 143个SSR标记和感病品种NorMan与抗病品种Linda杂交产生的300个F2群体构建了连锁图,最终鉴定出位于LG1、7和9上的3个白粉病抗性QTL,解释了97%的表型变异。杨学等[39]经培育得到亚麻抗白粉病品种9801-1。张倩[40]通过对121对InDel标记的筛选,成功获得了一个与亚麻抗白粉病基因紧密连锁的标记,其位于ChrNew02(第2条)染色体上,并将此基因暂时命名为pm-linum。除此以外,还有一些其他作物基于GWAS对白粉病进行研究,鲁宏伟[41]利用重测序的核心种质对黄瓜白粉病抗性基因进行GWAS,共检测到6个位点,其中pmG2.1、pmG5.2、pmG5.3被重复检测到,pmG1.1、pm G5.2、pmG5.3与前人获得的QTL定位结果相吻合。齐振宇[42]利用简化基因组测序和SNP分型技术,对甜瓜抗白粉病性状进行GWAS,最终将基因定位在2号染色体135976bp-1418829bp的位置。刘盼娜[43]利用核心种质进行重测序,对黄瓜茎蔓抗白粉病基因进行GWAS,共检测到4个抗性位点,pm-sG1.1、pm-sG2.1、pm-sG5.1、pm-sG5.2。
3 GWAS在其他作物中的应用
随着高通量测序技术的发展,水稻、玉米、大豆等参考基因组均已公布,这些物种中基因组范围的变异数据被用于遗传作图和作物演化的研究,亚麻在这方面的起步较晚,研究较少,以此为亚麻的品种改良和培育提供依据。白明兴等[44]以204份玉米自交系为关联群体,利用分布于玉米全基因组上的558529个SNPs对主要株型性状与产量的5个相关性状进行全基因组关联分析,利用Q+K模型检测到株高、穗位高、单穗重的13个显著的SNP位点,分别分布在第3、第5、第6、第7号染色体上,且在显著SNP上下游各50 kb范围共搜索到39个相关候选基因,其中包括12个有注释的基因。马娟等[45]为研究玉米出籽率的遗传机制,用309份玉米自交系作为关联群体,利用FarmCPU、CMLM和MLMM方法对玉米的出籽率进行全基因组关联分析,共鉴定出18个与出籽率显著关联的SNP和32个候选基因,其中包括5个主效SNP,共解释了0.067%~15.43%的出籽率变异率。Zhao等[46]为控制玉米中镉的积累,以269份玉米作为研究材料,利用43437个SNP对叶片中镉含量进行GWAS分析,最终鉴定出SYN25051为显著SNP,能够解释27.1%的表型变异,并验证了qLCd2为主效QTL,能够解释39.8%的表型变异。Ju等[47]利用224152个SNP,将重组自交系群体中的QTL定位与217个不同玉米品种的GWAS结合起来,研究玉米对枯萎病镰刀菌孢子的抵抗力,最终得到8个相关QTL和3个候选基因GRMZM2G0081223,AC213654.3_FG004 和 GRMZM2G099255。Chang等[48]利用62423个SNP标记对368个大豆育种品系的株高和主茎节数进行了GWAS,基于CMLM和mrMLM模型,CMLM检测出11个和13个分别与株高和主茎节数相关联的QTN;mrMLM检测出34个和30个与株高和主茎节数相关联的QTN。Fang等[49]收集了809种大豆种质资源,对84种农艺性状进行表型分析并进行了全基因组测序,最终揭示了23个基因座,包括已知的Dt1,E2,E1,Ln,Dt2,Fan和Fap以及16个未定义的相关基因座,对不同性状具有多效性效应。Li等[50]为研究大豆种子中油和蛋白质含量变化的遗传基础,利用1536个SNP对421份大豆品种种子的蛋白质含量和含油量进行GWAS,最终检测到与大豆种子的蛋白质含量和含油量相关的6个遗传位点。Zhang等[51]利用313份大豆种质的SNP标记对包括蛋白质、油脂、脂肪酸等种子组成相关性状进行GWAS,共鉴定出87个染色体区域与种子组成有关,解释了8%~89%的遗传变异,并在主要作用位点上发现了功能已知的候 选 基 因 GmSAT1,AK-HSDH,SACPD-C 和FAD3A。Zeng等[52]对283份大豆种质的33009个SNP标记对大豆耐盐性状进行GWAS,共检测到45个与大豆耐盐性显著相关的SNP。赵宏亮等[53]利用6704个SNP标记对261份籼稻核心种质资源的剑叶长、剑叶宽、剑叶叶面积、倒2叶长、倒2叶宽、倒2叶叶面积、每穗粒数、千粒重、单株产量的9个性状进行GWAS,共检测到68个相关联的QTL,其中QTgw5b的贡献率最大,为6.91%。Volante等[54]利用281份粳稻品种在缺水和多水条件下对种子形态、生理和产量相关等性状进行GWAS,最终得到3个与产量相关的位点,其中,Os05g0187500和Os04g0615000分别编码与谷粒重量相关的GW5和Nal1基因,Os04g0663600编码与腋芽形成相关的WOX1基因。
4 展望
全基因组关联分析技术虽然具有检测范围广、精确度高等优势,但也不能完全替代传统的QTL作图方法,基因之间、基因与环境的互作都会影响GWAS的分析结果。其次,GWAS只能粗略预测候选基因的区段,后续还要结合其他的实验方法来精确定位候选基因。虽然GWAS技术存在不足,但随着技术的发展,这些问题最终会得以解决。同时,GWAS也将在亚麻如下几个方面中加以探讨研究:
(1)GWAS应用于亚麻重要性状研究的具体方法和策略还处于不断探索和改进的阶段,尤其相较于水稻、玉米、大豆等作物,因此,未来可多参考这些作物利用GWAS对亚麻产量相关性状、品质相关性状、抗逆性以及其他性状进行研究。
(2)GWAS在亚麻遗传学研究上取得了一定的进展,但主要集中在产量相关性状和品质相关性状,关于抗病的研究相对较少,亚麻白粉病、枯萎病、斑枯病等,不仅影响亚麻的品质,也会影响亚麻的产量,所以利用GWAS对亚麻抗病的遗传机理进行分析已经成为刻不容缓的工作。
