植物油中多环芳烃的测定及方法验证
——高效液相色谱法(荧光检测器)
2021-09-01赵雪峰曹静杰贾松涛郭倩倩李梦雨赵林萍
◎ 李 超,赵雪峰,陶 燕,曹静杰,贾松涛,郭倩倩,李梦雨,赵林萍
(河南中标检测服务有限公司,河南 郑州 450001)
植物脂肪是人类饮食的重要组成部分,是能量和必需脂肪酸的来源,也是脂溶性维生素的载体。油籽、植物油和脂肪制品等脂肪含量高的食物易受多环芳烃(PAHs)的污染,多环芳烃具有亲脂性,因此在动植物的脂肪细胞中易于积累。植物油可能会被多环芳烃污染,如在油品加工过程使用了被污染的萃取溶剂。在一定程度上,油品的污染还可能与环境污染有关。在大气存在多环芳烃污染的情况下,由于降水,植物在生长季节可能发生表面污染,这种污染可能会被转移到最终产品中。许多研究人员发现,同一种油在不同生产批次中的多环芳烃含量存在显著差异[1-2]。
世界卫生组织国际化学品安全规划署(IPCS)、欧盟委员会粮食科学委员会以及粮农组织/世卫组织食品添加剂专家委员会分别于1998年、2002年和2005年对多环芳烃进行了评估。国际癌症研究机构将苯并(a)芘归为第一类致癌物,而将苯并(a)蒽、苯并(b)荧蒽和归为2B类致癌物(可能对人类致癌的化合物)[3]。
近年来,植物油受到越来越多的消费者青睐,各种菜籽油、葵花籽油、大豆和橄榄油及其混合物产品出现在市场上。研究者越来越关注对多环芳烃遗传毒性、诱变性和致癌性进行研究,为保障食品安全,需要不断监测食品中多环芳烃的含量,以持续监测安全风险。
本文对高效液相色谱法测定多种多环芳烃的方法进行验证,进而研究了市场上不同种类植物油中多环芳烃的污染程度,包括苯并(a)芘(BaP)、苯并(a)蒽(BaA)、苯并(b)荧蒽(BbFA)和䓛(CHR)等化合物含量的总和,为国家相关部门制定这些污染物质在食品中的限量值提供数据基础。
1 材料和方法
1.1 试剂和样品
乙腈、环己烷、石油醚、甲醇,均为色谱纯;氯化钠、氢氧化钾、无水亚硫酸钠(分析纯);符合实验室用水标准中的一级水;氮气;中性氧化铝净化柱;多环芳烃标准混合溶液;质控样为橄榄油CRM。
市售的30种菜籽油样品、10种葵花籽油样品和12种其他油样品包括油橄榄果渣油、橄榄油菜籽油调和油、大豆油以及椰子油。菜籽油样本包括普通精制成品油、精制初榨菜籽油和冷过滤精制初榨菜籽油,分别为国产(12个样品)和进口(18个样品)。分析前,样品根据生产厂家的建议进行存储。液体样品于室温放置2 h,固体样品置于60 ℃水浴中融化。
1.2 仪器与设备
戴安UltiMate 3000高效液相色谱仪(配荧光检测器);PAH C18反相键合固定相色谱柱(250 mm×4.6 mm,5 μm)。
2 分析步骤
2.1 样品净化
为了使多环芳烃萃取物的纯度足够用于色谱分析,采用氧化铝填充玻璃柱进行纯化。将氧化铝与水的混合物(9∶1,w/w)倒入装有多孔玻璃的玻璃柱中,在氧化铝填料上填充无水亚硫酸钠干燥剂。通过石油醚对净化柱进行调节后,用10 mL石油醚稀释0.4 g的油脂样品,将样品移入氧化铝柱中,用60 mL石油醚洗脱样品,流速为1 mL·min-1,弃去最初的20 mL石油醚,收集余下流出物。流出物经旋转蒸发仪浓缩,氮吹除去溶剂至近干,用乙腈定容至1 mL,混匀,过0.22 μm有机相微孔滤膜,待色谱仪检测。
2.2 高效液相色谱分析
用自动进样器将20 μL样品注入高效液相色谱中。柱温保持在35 ℃,流动相为乙腈和水。梯度洗脱时间见表1,流速为1 mL·min-1。激发和发射(Ex/Em)波长:BaA和CHR为260 nm/420 nm、BbFA和BaP为290 nm/430 nm。

表1 梯度洗脱时间表
2.3 校准曲线的制备
采用外标法建立校准曲线。使用多环芳烃标准混合溶液配制标准中间工作液,浓度为10 ng·mL-1,分别吸取一定量的标准中间工作液,经稀释后配制成浓 度 点 为0.20 ng·mL-1、0.80 ng·mL-1、4.00 ng·mL-1、8.00 ng·mL-1及10.00 ng·mL-1的系列标准溶液,以峰面积为纵坐标、浓度为横坐标建立标准曲线。
2.4 样品分析
质量控制过程包括对盲样和作为质控样的有证标准样品进行检测。
3 统计分析
3.1 测定方法的线性工作范围
对每条多环芳烃校准曲线计算浓度限度的变异系数,然后在0.05显著水平下使用F-Snedecor检验对变异系数进行检验,同时计算相关系数。检出限(LOD)和定量限(LOQ)的计算公式如式(1)和式(2)所示。该方法的灵敏度以校准曲线的斜率为准。
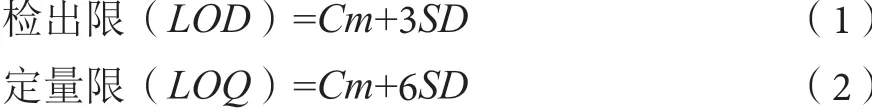
式中:Cm-低浓度样本中多环芳烃的平均浓度;SD-标准偏差。
3.2 统计结果分析
验证过程中得到的结果通过Dixon Q检验进行识别和剔除异常值,然后对每个多环芳烃浓度计算下列统计参数,包括重现因子(正确性)、方差、标准差、变异系数、标准不确定度和扩展不确定度、重复性的置信区间和相对标准偏差。
4 结果与讨论
检出限(LOD)、定量限(LOQ)和回收率均符合国家标准《合格评定 化学分析方法确认和验证指南》(GB/T 27417—2017),在0.20~10.00 ng·mL-1浓度范围内,各物质的校准曲线均呈良好的线性关系,相关系数均大于0.998。这些曲线用于检验该方法在0.80 ng·mL-1和8.00 ng·mL-1两种浓度下的重现性和精密度。相关测定结果见表2。根据表2可以看出该方法适用于测定植物油中4种多环芳烃含量总和。
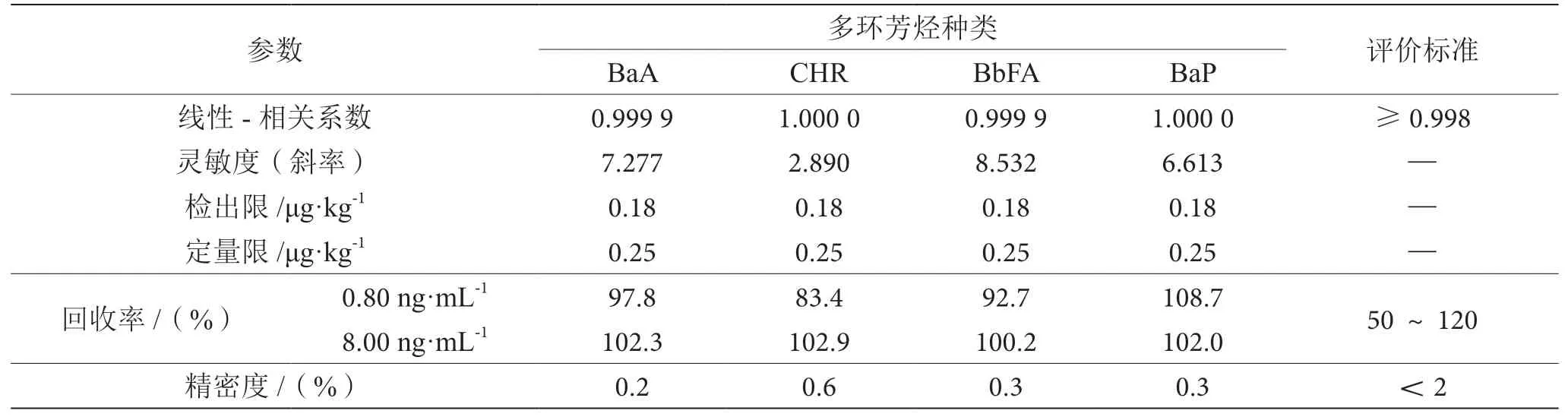
表2 方法验证参数和评价标准表
标准混合溶液和菜籽油中多环芳烃的色谱图分别见图1和图2。
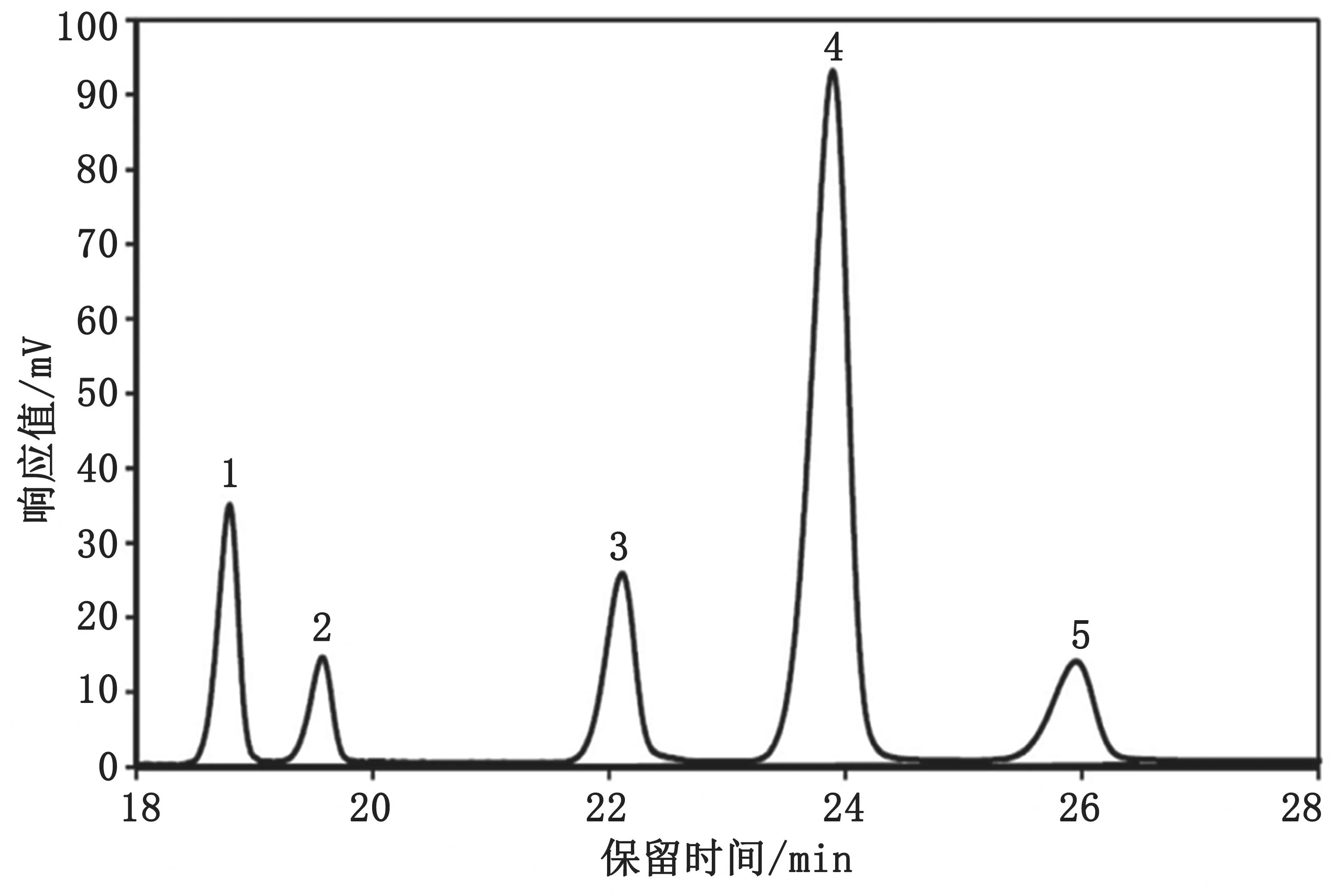
图1 多环芳烃标准混合溶液色谱图
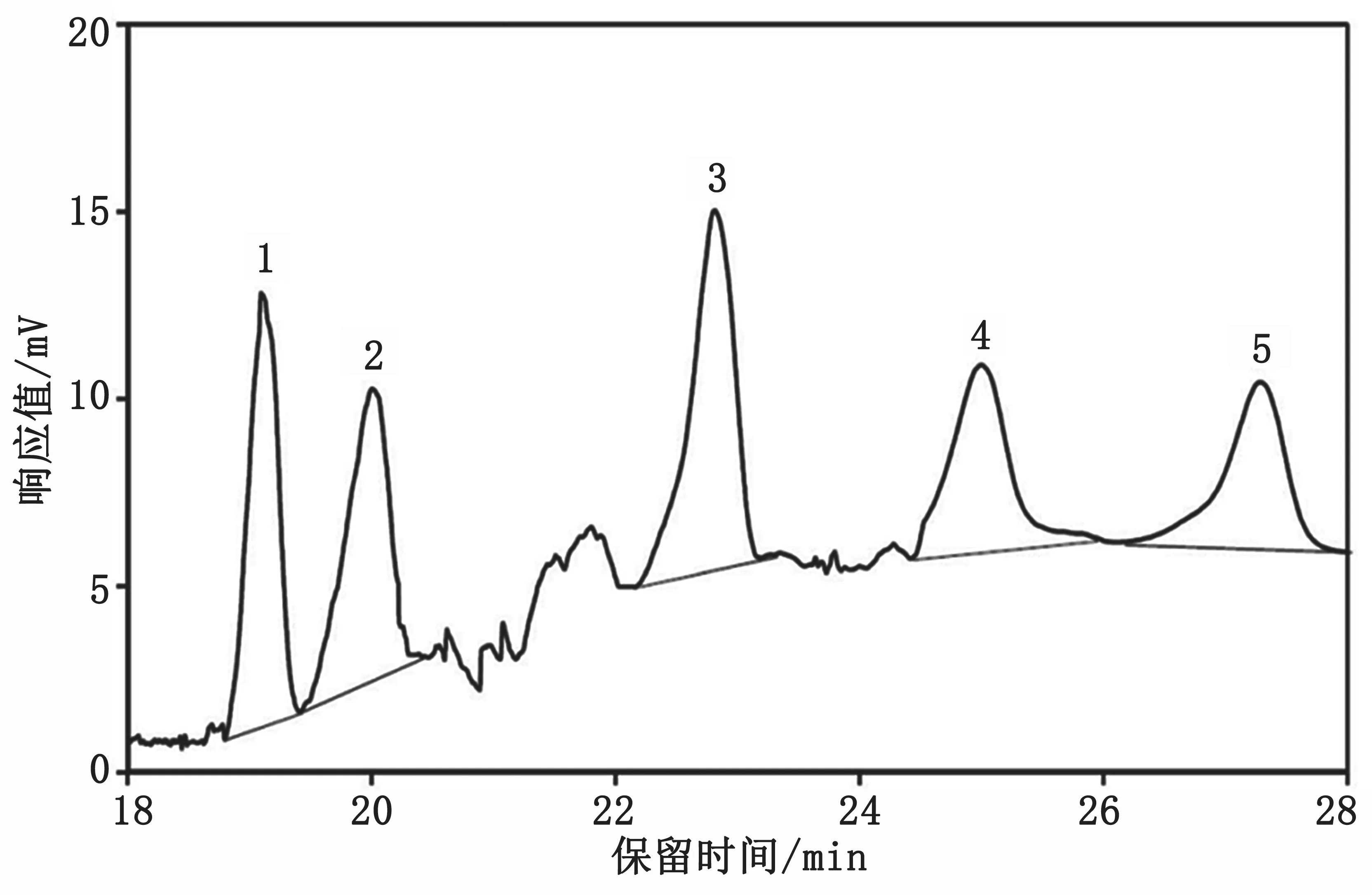
图2 菜籽油中多环芳烃的色谱图
菜籽油中测定的苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽、䓛以及这4种多环芳烃的含量总和见表3,苯并(a)芘、苯并(a)蒽、䓛和苯并(b)荧蒽的回收率分别在80%~103%、87%~109%、87%~108%和91%~110%范围内。精制菜籽油中4种多环芳烃的含量最大值为1.30~2.40 μg·kg-1,精制初榨菜籽油中4种多环芳烃的含量最大值为1.10~1.95 μg·kg-1,考虑到不确定度,含量差别并不大。冷过滤精制初榨菜籽油中苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽和的含量均低于1 μg·kg-1,4种PAHs含量之和(2.91±0.58) μg·kg-1也显著低于精制菜籽油和精制初榨菜籽油。
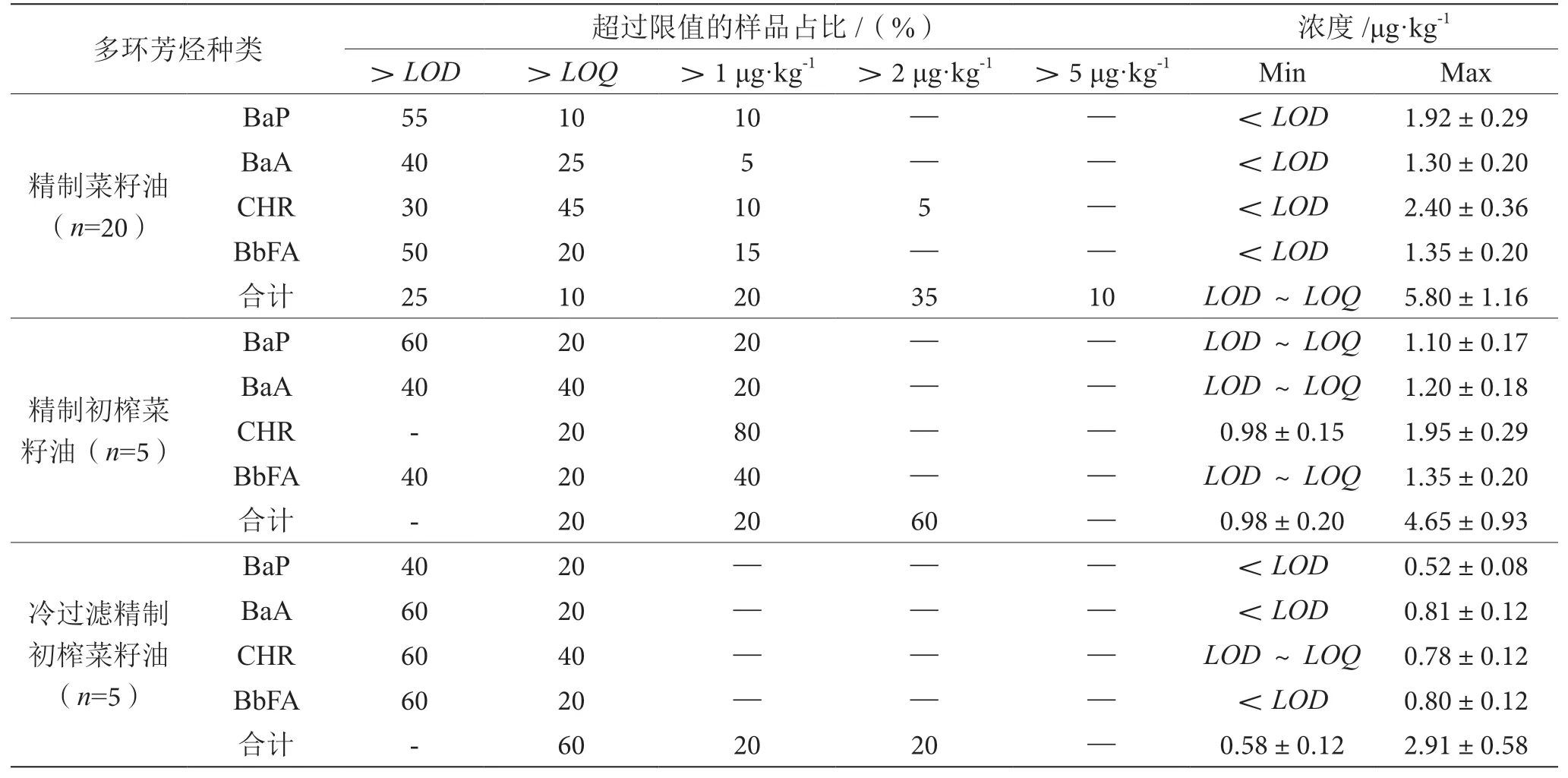
表3 菜籽油样品中多环芳烃含量表
在菜籽油中,含量占比最大的多环芳烃化合物为,其中3个菜籽油样品中䓛含量超过1 μg·kg-1,4个精制初榨菜籽油样品中其含量超过1 μg·kg-1。这一结果与ALOMIRAH等[4]的研究结果一致,其研究团队报道在83%的植物油样品中检测出了䓛。
所有样品中4种多环芳烃含量之和均不超过10 μg·kg-1,这 与CIECIERSKA和OBIEDZI SKI的报道一致。CIECIERSKA和OBIEDZI SKI研究发现精炼工艺在油籽加工过程中起着关键作用,因为其显著降低了产品中的多环芳烃含量。菜籽油中苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽和䓛的污染水平,与其他国家测定的精制植物油中这些化合物的平均水平相当[5]。
在检测的精制葵花籽油中苯并(a)芘含量不超过2.0 μg·kg-1,30%的样品中苯并(a)芘含量低于方法验证时测定的检出限(0.18 μg·kg-1),50%的样品中苯并(a)芘含量在0.18~0.25 μg·kg-1。在大多数精制葵花籽油样品中BaP、CHR和BbFA含量均低于定量限(分别是80%、70%和80%)。多环芳烃含量最高的油品中分别含有BaP(1.86±0.28)μg·kg-1、BaA(0.92±0.14)μg·kg-1、CHR(1.42±0.21)μg·kg-1、BbFA(1.58±0.24)μg·kg-1。许多研究[1,5]表明食用油通过精制过程,特别是使用混合吸附剂(活化漂白土和活性炭),多环芳烃的含量会降低。
油橄榄果渣油和添加橄榄油的菜籽调和油样品中多环芳烃含量均适中,苯并(a)芘最小含量低于检出限,而最大含量分别达到0.25 μg·kg-1和0.45 μg·kg-1。油橄榄果渣油中4种多环芳烃含量总和与添加橄榄油的菜籽调和油中含量之间没有显著差异,具体结果见表。有研究认为,多环芳烃吸附在果实表面,在萃取过程中转移到油脂中,油橄榄果渣油可能是多环芳烃的重要来源。多环芳烃含量可作为确定不同植物油品质和优化精制工艺的重要指标。
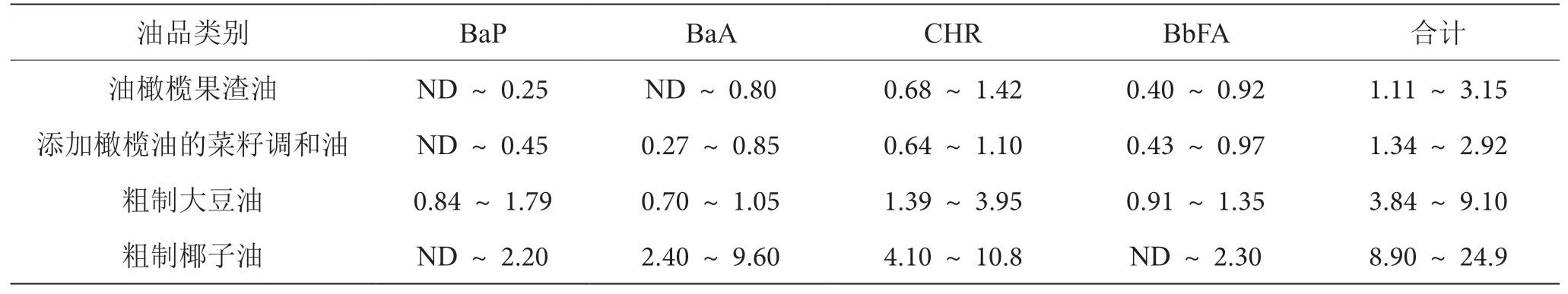
表4 其他种类植物油中PAHs含量表(单位:μg·kg-1)
粗制大豆油中苯并(a)芘和4种多环芳烃含量之和均显著高于精制成品油,含量之和最高为(9.10±1.82)μg·kg-1。YE等[6]在粗制大豆油和精制大豆油中检测到的PAHs含量明显较低。他们认为由于油经过了中和、漂白和脱臭过程,使得苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽的含量降低。未精制的椰子油中多环芳烃的污染最为严重。
在本研究中,苯并(a)芘含量与4种多环芳烃含量之和呈现显著的线性相关(r=0.947)。不同种类食用植物油样品中,85.8%的样品苯并(a)芘含量<2 μg·kg-1,此外在测定的多环芳烃中,䓛的占比最大。
5 结论
所有被测植物油样本中含有的苯并(a)芘及苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽、䓛(4种多环芳烃的总和)的含量均低于国外标准规定的限值[7]。与成品油相比,非成品油的PAHs污染水平明显更高;未精制的椰子油中多环芳烃含量最高;精制菜籽油和精制初榨菜籽油中4种PAHs含量无显著差异,而冷过滤精制初榨菜籽油中各PAHs的最高含量均显著低于其他菜籽油。
