邻苯二甲酸衍生物构筑的双核钴(II)混配配合物的结构和磁性研究
2021-08-31王爱胡子佳袁彩霞
王爱,胡子佳,袁彩霞
(山西大学 分子科学研究所,山西 太原 030006)
0 引言
近年来,过渡金属配合物在分子磁体、气体吸附、荧光、新型材料、分子识别、光电效应、非线性光学性能以及多相催化等诸多领域显示出诱人的应用前景[1-4]。作为强磁性材料的一个分支,无机磁性材料的特殊性能得到了人们的广泛关注,然而该种材料存在加工难度大等缺陷。为了解决这一问题,金属有机磁性材料出现在人们的视野中。过渡金属配合物分子磁体作为一类新型功能材料,是通过化学方法将自由基或顺磁离子(主要指过渡金属离子)及抗磁配体以自发组装和控制组装的方式组合而成的磁性化合物。分子的磁性一般分为抗磁、顺磁、铁磁、反铁磁与亚铁磁性。而磁性的来源为组成物质的分子中的电子轨道磁矩、电子自旋磁矩和核自旋磁矩在空间的取向及其磁矩间的相互作用。不同的是,分子磁性材料有着密度小、透明度高、易于加工等优点,有望在航天材料、微波材料、信息记录材料、光磁及电磁材料等方面加以应用。因此,构建结构稳定、性能优异的分子磁性材料已经成为分子磁学领域研究的热点问题。特别是一定条件下,第四周期3dn(其中4 ≤n≤7)型的过渡金属配合物呈现出与光电转换能力密切相关的自旋转换能力,可作为一种具有潜在应用价值的新型信息存储材料[5]。特别是二价钴配合物因其独特的电子自旋行为和潜在应用价值而得到人们的重视[6-10],例如4,4′-联吡啶和Co(II)形成的配位聚合物,其变温磁化率实验值表明,该化合物中钴离子之间存在着较弱的反铁磁性,且有自旋转换的倾向。进一步对其表面光电压谱测试。在紫外-可见光照射下,该配位聚合物表面出现的光伏响应带实验结果表明其明显的光电转换性能[6]。这种引入辅助配体的合成方法,不仅能够有效提高金属有机配合物的结构多样性;同时也极大地丰富了潜在的磁性材料分子库中的过渡金属配合物。
设计和合成功能型过渡金属配合物过程中,配体的选择发挥着至关重要的作用。本项工作选择的配体为一个非对称的含氮芳香多羧酸类配体,其中含氮杂环为三氮唑基团。其结构特征为:a)配位点多,含有三个取代芳香环的羧基和三氮唑上的N原子都可以作为有效配位点,提高了配位模式的多样性;b)含有苯环和三氮唑两个芳香环,使电子可以在形成的配合物内部进行有效传递,有望得到一些在光、电、磁、及催化等方面具有优良性能的化合物;c)亚甲基的存在进一步增加配体在形成配合物时,金属与配体连接方式的多样性。苯环与三氮唑通过亚甲基相连,在形成固态晶体的过程中,根据空间位阻的影响形成不同结构类型的配合物。由该配体构成的一维单核锰配位聚合物中锰(II)间呈现弱的铁磁相互作用(外斯常数θ值为0.082 K),可作为分子磁性材料。[11]
研究表明,对于过渡金属配合物形成的分子磁性材料,金属阳离子作为自旋载体通过短的桥联配体与配体未配对电子连接时,分子轨道有较大的重叠,确保了金属离子间有效的耦合。在构建多核超分子磁体时,简单的羧酸、较小的酮或较大的多齿螯合配体可以作为有效的桥梁。其中多羧酸配体采用可变的配位模式,可以通过一个或两个氧原子桥接金属阳离子,从而促进配合物中结构的有效延伸和磁相互作用[12]。然而,目前单链磁体或具有磁性的低维度的离散结构单元的构建还极具挑战。选择钴离子作为自旋中心的主要原因为:金属阳离子在配合物中不仅由于表现出多种配位模式,并且根据其氧化状态,有望合成具有高磁各向异性的磁性分子材料。在钴配合物中,较常见的Co2+(电子组态为3d7),罕见的Co4+(3d5)与顺磁性有关[13],而常见的Co3+离子通常采用低自旋3d6构型,具有抗磁性[14]。
本文我们采用水热合成的方法,得到了邻菲罗啉和5-((3-羧基-1,2,4-三唑-1-基)甲基)-邻苯二甲酸(H3L)混配的零维双核钴(Ⅱ)配合物。通过X-射线单晶衍射实验解析其固态晶体结构,利用红外光谱、热重分析、粉末衍射图样等手段对其结构及性质进行表征。通过对其磁化率随温度变化情况的分析,进一步探讨了零维双核钴配合物结构与其反铁磁性之间的构效关系。研究表明:含有零场分裂作用的单核Co(II)配合物在2 K~200 K 的温度变化范围内,钴离子表现出明显的单轴各向异性。
1 实验部分
1.1 实验试剂和仪器
分析纯的氯化钴(CoCl2·6H2O)来自天津市北辰方正试剂厂,邻菲罗啉为北京芳草医药化工研制公司分析纯试剂;5-((3-羧基-1,2,4-三唑-1-基)甲基)-邻苯二甲酸(H3L)购置于山东省济南市恒化试剂股份有限公司。其它化学试剂均为分析纯级(AR)。FT-IR 仪(Bruker Tensor 27 型傅立叶红外光谱仪);TGA/SDTA 851e 仪(瑞士Meltter-Toludo 公司);单晶衍射仪(德国BIUKER 公司APEX CCD II 型);X-射线粉末衍射仪(Bruker D8,λ=1.540 598 Å,粉末衍射仪);磁 强 度 计(SQUID Quantum Desigh Model MPMS-7 型)。
1.2 [Co2(phen)2(HL)2·(H2O)4](配合物1 的合成)
将CoCl2·6H2O(47.50 mg,0.20 mmol),H3L(38.00 mg,0.20 mmol),10.0 mL H2O 和5.0 mL甲醇逐一加入到25 mL 聚四氟乙烯内衬管中,搅拌均匀后再将其放入不锈钢反应釜中。反应釜在140°C 的烘箱内持续反应72 h 后,自然冷却降至室温。管壁四周和管底中均匀分布有粉棕色薄片状晶体。再经过在显微镜下筛选、过滤,并用蒸馏水反复清洗后,将样品放置与真空干燥器干燥(产率:67%)。IR 数 据(cm-1,KBr):3 456(m),3 148(w),1 715(m),1 632(m),1 550(w),1 483(m),1 461(w),1 417(s),1 361(m),1 253(s),1 240(w),1 209(s),1 193(m),948(w),858(s),752(m),667(m)(s:strong;m:medium;w:weak)。
2 结果与讨论
2.1 [Co2(phen)2(HL)2·(H2O)4](配合物1 的晶体结构表征)
室温环境下,配合物1 的X 射线单晶衍射数据在Bruker D8 venture 衍射仪上进行收集,特征波长为0.710 73 Å 的钼靶Mo-Kα为单晶数据收集的辐射光源。确定其晶胞参数,数据还原和吸收校正的程 序 分 别 为SMART,SAINT 和SADABS[15]。晶体的初始结构利用SHELXS-2014 程序中的直接法进行解析[16],并用最小二乘法对初始结构进行逐步精修。在所有非H 原子的热振动参数精修之后,根据C 原子的杂化理论对其加氢,其中C-H=0.93 Å,Uiso(H)=1.2 Ueq(C)。相关的晶体学数据和结构精修重要参数呈现于表1。配合物中部分键长及键角列于表2。

表1 配合物1的晶体学数据和结构精修参数Table 1 Crystallographic data and structure refinement data of complex 1

表2 配合物1中部分键长、键角信息(Å,º)Table 2 Selected bond lengths(Å)and bond angles(º)of complex 1
X 射线单晶衍射结果表明,双核钴配合物(1)结晶为单斜晶系,空间群为P21/c。如图1(a)所示,配合物1 的不对称单元结构包含一个钴离子,一个不完全去质子的主配体(HL2-),一个邻菲罗啉(phen)分子和两个游离的水分子。中心金属离子Co2+形成了六配位的几何构型[CoN3O3],其中两个N 原子来自邻菲罗啉,第三个N 原子来源于主配体三氮唑基团,三个O 原子来自于两个不同的羧基。 Co-O键的键长范围在2.073 Å ~2.180 Å 之间,Co-N 键的键长范围在2.106 到2.119 Å 之间。[17]O-Co-O键的夹角范围在60.73°~159.38°之间。O-Co-N键的夹角范围在79.63°~169.49°。[CoN3O3]配位几何呈现畸变的八面体构型。 两个相邻的[CoN3O3]通过不完全脱质子的主配体(HL2-)桥连形成双核钴零维结构(如图1b),其中配体通过μ2-ƞ2:ƞ1:κ-O1:O2:O3:N3的配位模式连接两个距离为9.406 Å 的Co(II)离子。如图1c 所示,主配体中的苯环Cg1 环和Cg1i环之间存在较短的π…π 相互作用(Cg1 环是由C14、C15、C16、C17、C18、C19 组成;对称代码:i = -x+2,-y+1,-z+1)。相邻的Cg1环和Cg1i环之间距离为3.906 Å。结合图1c 和1d,我们发现配体未脱质子的羧基与相邻的羧基形成晶体中最短的分子间氢键(其中氢键的给体O6 与受体O2vii的距离为:2.658 Å)。晶胞中游离的水分子与主配体中未配位的羧基氧原子形成三种类型的分子间氢键。氢键的给体和受体距离分别为:2.793(O8···O4i,i =x,y+1,z+1),2.811(O8···O4ii,ii =x,-y+1/2,z+1/2)和2.884 Å(O8···O3ii,ii =x,-y+1/2,z+1/2)。此外,相邻的两个游离水分子之间也形成了分子间氢键(O7···O8:2.811 Å)。由此可见,π-π 堆积和分子间较强的氢键作用,使配合物1 呈现三维网状结构。

图1 (a)配合物1 的配位环境图;(b)配合物1 的双核钴环状结构;(c)配合物1 的π…π 相互作用示意图;(d)由分子间氢键(蓝色色虚线)构筑而成的三维网络结构图Fig. 1 (a)The coordination environment diagram of complex 1;symmetric code:(i)-x+2,-y+1,-z+1;(b)Dinuclear Co(II)structure of complex 1;(c)The π…π interactions of complex 1;(d)The 3D structure of complex 1 constructed by inter-molecular hydrogen bonds(blue dashed line)
2.2 [Co2(phen)2(HL)2·(H2O)4](配合物1 的热重和红外谱图分析)
热重分析实验以10 ℃/min 的速度将配合物1从室温升温到800 ℃来测试其热稳定性。如图2(a)所示,在25 ℃~180 ℃范围内配合物1 失重约6.34%,这是由于脱去了晶胞内的四个游离水分子导致(理论值为6.38%),剩余的配合物1 在300 ℃~800 ℃的温度范围内逐渐分解,最终产物为氧化钴。
配合物1、主配体H3L 以及辅配体邻菲罗啉(phen)的红外光谱图如图2(b)所示。配体H3L 中羧酸的-OH 基团的伸缩振动为3 456 cm-1处的宽峰。配合物中游离的水分子为3 462 cm-1的宽峰。主 配 体 上 羧 基 的 特 征 峰 从 波 数 为1 715 和1 637 cm-1分别移动到较低波数1 712 和1 632 cm-1。与中性配体上羰基的特征峰相比较,配合物1中的羧基特征峰发生了红移,进一步证实金属离子与配体的羧基发生配位。同时,第二配体的C-N键的特征峰从1 589 cm-1移动到了1 550 cm-1处,明显的位移证明配合物中N 原子参与了配位。

图2 (a)配合物1 的热重分析图谱;(b)配合物1,配体H3L 和邻菲罗啉(phen)的红外图谱Fig. 2 (a)The TGA of complex 1;(b)The IR spectra of complex 1,H3L and phen
2.3 [Co2(phen)2(HL)2·(H2O)4](配合物1 的粉末衍射图样)
由图3 可知,配合物1 的单晶模拟衍射图样与其粉末样品的衍射实验值基本吻合,表明产物配合物1 为纯相。为后续单晶样品变温磁化率的测试提供必要的实验基础。

图3 配合物1 单晶结构模拟衍射图样(红色)和实验测得的配合物1 粉末衍射图样(黑色)对比Fig. 3 PXRD of experimental(black)and simulation(red)for complex 1
2.4 [Co2(phen)2(HL)2·(H2O)4](配合物1 的磁性分析)

表3 配合物1中氢键的几何信息(Å,º)Table 3 Hydrogen-bond geometries(Å,º)for 1
配合物1 在2 K ~300 K 范围内的变温磁化率如图4 所示。在300 K 时,配合物1 的摩尔磁化率(χm)与温度(T)的乘积χmT 的实验值是4.12 cm3·K·mol–1,比 两 个 孤 立 自 旋 态 的Co(II)的 理 论 值3.75 cm3·K·mol-1(S=3/2,g=2)略高。随着温度的降低,χmT 逐渐增大。从图中我们看出,χmT 在204 K 时达到 最大值4.73 cm3·K·mol-1,随着温 度的持续降低呈现逐渐下降的趋势。当温度降至2 K时,χmT 达到最小值1.95 cm3·K·mol-1,说明配合物1 中存在着反铁磁作用。在2 K~300 K 范围内,磁化率随温度的变化规律遵循居里韦斯定律,其中居里 常 数C = 5.07 cm3·K·mol-1,外 斯 常 数θ =-20.71 K 小于0,进一步验证了配合物1 中二价钴离子具有反铁磁耦合作用。

图4 配合物1 中磁化率、磁化率与温度的乘积和磁化率的倒数曲线(红色细线为公式1 和2 的最佳拟合值)Fig. 4 The diagram of χM,χMT and χM-1 of complex 1(the red solid lines represent the best fittings)
尽管配合物1 中两个Co(II)离子之间通过配体HL2-桥连。但是与其它双核钴(II)结构不同的是,该配合物中Co(II)…Co(II)之间的距离为9.406 Å。相邻钴离子较远的距离会导致金属离子之间的相互作用减弱,进而导致常用的双核钴磁化率公式不能很好的拟合配合物1 的变温磁化率曲线。因此,我们采用单核Co(II)离子模型,并考虑单核金属离子的零场分裂作用(参数用D 表示)来拟合配合物1[18-19]实验测得的变温磁化率曲线,并进一步分析和解释配合物磁耦合作用随温度的变化情况。含有零场分裂作用的单核Co(II)离子磁化率表达式如下:
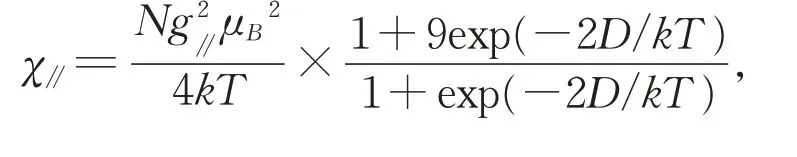

根据公式(1 和(2),配合物在2 K~200 K 的温度范围内,磁化率随温度变化曲线的最佳拟合值为:g//= 2.48(5),g⊥= 1.81(8),g= 2.03(8),D=-54.32(2)cm-1,R= 4.22(2)× 10-4。由零场分裂作用所引起的负的D 值进一步表明Co(II)离子的单轴磁各向异性。[20-24]
3 结论
本文采用H3L(5-((3-羧基-1,2,4-三唑-1-基)甲基)-邻苯二甲酸)为主配体,以邻菲罗啉为辅助配体,与Co(II)离子通过水热的方法成功合成了一种新型的钴配合物1。它是由金属钴、主配体H3L和phen 构筑的一个零维双核钴结构,其配体采用μ2-ƞ2:ƞ1:κ-O1:O2:O3:N 的配位模式连接两个相邻的Co(II)离子。配合物1 的磁性分析结果表明,配合物1 具有反铁磁性。进一步磁性拟合数据分析表明,该单核钴模型中存在着零场分裂效应以及单轴磁各向异性。本项单核钴模型用于解释零维双核钴结构的磁化率随温度变化情况的研究,为过渡金属配合物磁性材料的潜在应用提供一定的实验基础。
