Bacterial community diversity of meltwater runoff and soil in Midre Lovénbreen glacier in Ny-Ålesund, Arctic
2021-08-18LINLidongWANGNengfeiHANWenbingZHANGBotaoZANGJiayeLIQinxinQinYilingWangLongZhangFangLiuJie
LIN Lidong, WANG Nengfei, HAN Wenbing, ZHANG Botao, ZANG Jiaye, LI Qinxin, Qin Yiling, Wang Long, Zhang Fang & Liu Jie
Bacterial community diversity of meltwater runoff and soil in Midre Lovénbreen glacier in Ny-Ålesund, Arctic
LIN Lidong1, WANG Nengfei2*, HAN Wenbing2, ZHANG Botao1, ZANG Jiaye2, LI Qinxin1, Qin Yiling2, Wang Long3, Zhang Fang4& Liu Jie3
1College of Chemistry and Chemical Engineering, Qingdao University, Qingdao 266071, China;2First Institute of Oceanography, Ministry of Natural Resources, Qingdao 266061, China;3Department of Bioengineering, College of Marine Sciences and Biological Engineering, Qingdao University of Science & Technology, Qingdao 266042, China;4Polar Research Institute of China, Shanghai 200136, China
Glacial meltwater runoff is a dynamic ecosystem. On the one hand, nutrient concentration changes as it flows from upstream to downstream, and on the other hand, bacterial community structure changes due to its contact with nearby soil during the flow process. We studied meltwater and soil in the Midre Lovénbreen glacier region, to explore changes in bacterial diversity as meltwater flows, and the relationship between meltwater and soil bacterial diversity. As glacial meltwater flows from upstream to downstream, the relative abundance of dominant bacterial groups changes. In addition, we found that during the flowing process, nutrient exchange and bacterial contact had occurred between the meltwater runoff and the soil. As a result, the distribution patterns of some bacteria in the meltwater are very similar to those in the soil. Finally, we combined distance-based redundancy analysis and weighted correlation network analysis to show that NO3−-N and NO2−-N are the most two significant factors affecting glacial meltwater and soil, respectively. Our results suggest that in such a close-knit ecosystem, the interaction of glacial meltwater with soil, as well as environmental factors, together determine bacterial community composition.
Arctic, glacier meltwater, bacterial diversity, high-throughput sequencing
1 Introduction
Svalbard’s glaciers and ice sheets account for about 6% of the world’s glaciers outside the Greenland and Antarctic ice sheets (Farinotti et al., 2019). The Arctic has warmed twice as much as the global average over the past 20 years due to global warming, and the Svalbard glacier is on the edge of retreating Arctic sea ice, making it one of the fastest-warming regions on Earth (Noël et al., 2020). In the last few decades, researchers have come up with the concept of glacial ecology (Hodson et al., 2008). Bacteria were the dominant microorganisms in the glacial niche, followed by fungi and algae (Anesio et al., 2017). Glaciers at different geographical locations show significant differences in microbial community structure (Liu et al., 2009; Zhang et al., 2009), controlled by several environmental and climatic drivers (Takeuchi and Kohshima, 2004; Bhatia et al., 2006). Therefore, it can be said that microorganisms are sensitive indicators of climate change and a component of biogeochemistry (Wang et al., 2015; Guo et al., 2018). In addition, subglacial microorganisms play an essential role in driving biogeochemical cycles of carbon, nitrogen and phosphorus in the High Arctic (Wang et al., 2016).
Glacial meltwater, as a unique environment for prokaryotes and eukaryotes, is regarded as a true microbial community (Anesio and Laybourn-Parry, 2012). Microbes in these ecosystems can survive and reproduce at low temperatures, which means they have overcome a key hurdle inherent in a prolonged cold environment. The glacial meltwater was found to be the home of a number of bacteria that play an important role in global microbial activity and nutrient cycling (Anesio et al., 2017). And this kind of water may be a valuable source of the glacial microbiome released from englacial and subglacial environment (Sułowicz et al., 2020).Kohler et al. (2020) found that Proteobacteria, Bacteroidetes and Actinobacteria were all the bacteria with high abundance in the outlet bacterial communities of glacial melt water in the Arctic region. The research of Fodelianakis et al. (2022) showed that glacier-fed streams, as an extreme and rapidly disappearing ecosystem, had diverse microbial communities. Sułowicz et al. (2020) assessed the microbial community structure of a subglacial drainage system in Svalbard and showed that the nature of water at different locations determines bacterial diversity. However, we don’t know enough about how bacterial communities change with the dynamics of glacial melt water. In glacial habitats, when temperatures are high, upstream glacial runoff firstly interacts with glacial soils and then flows into lakes or streams (Yang et al., 2016). The soil is connected to lakes and streams, and is an essential microbial niche in glacial ecosystems, regulating biogeochemical cycles. As glacier retreat continues, glacial runoff flows downstream into estuaries, thereby diverting nutrients and microbial communities from glacial meltwater into the ocean (Sajjad et al., 2020). In this process, we believe that it is essential to fully understand the bacterial distribution in soil and meltwater runoff ecosystems under accelerated glacier retreat.
Glacial meltwater connects discrete cryosphere habitats to downstream marine ecosystems (Milner et al., 2017). In addition to exporting fresh water, glaciers subsidize microbial productivity by transporting dissolved matter and nutrients through meltwater runoff (Kohler et al., 2017; Hatton et al., 2019). Meltwater runoff also exchanges nutrients with the surrounding glacial soil. But Sajjad et al. (2021) believed that the bacterial diversity of soil samples in glacial areas was significantly affected by ecological parameters more than that of meltwater runoff. We wanted to explore changes in bacterial diversity as glacial meltwater flows and the interaction between meltwater and soil. Therefore, we used high-throughput sequencing to evaluate the glacial runoff and adjacent soils of the Midre Lovénbreen glacier in Svalbard, as well as the seawater at the downstream estuary. This study mainly solves the following problems: (1) What are the differences in bacterial community composition between the upper and lower reaches of meltwater Midre Lovénbreen glacier? (2) What are the differences in bacterial community composition between meltwater of Midre Lovénbreen glacier and nearby soil? (3) What are the key geochemical factors that determine the composition of the bacterial community in the the Midre Lovénbreen glacier runoff and soil?
2 Materials and methods
2.1 Study sites and sample collection
Midre Lovénbreen glacier is located in the Konsfjord region of Svalbard (78°53′N, 12°02′E), in the Arctic. The glacier is about 6 km long, covers an area of about 5.5 km2and has a maximum thickness of about 180 m (Björnsson et al., 1996). The altitude of the glacier at the snout is about 50 m and increases to about 600 m above sea level at the head wall (Reddy et al., 2009). The distance from the mouth of the glacier to the point where meltwater flows into the ocean is about 2–3 km.
Sampling sites are shown in Figure 1, we set up 11 sampling sites along the glacier runoff from upstream to downstream. Among them, eight sampling points collected water samples from glacial runoff (SR01-SR08), SR01-SR03 is the upstream of glacial meltwater, and SR04-SR08 is the downstream of glacial meltwater, and soil near glacial meltwater was collected at three sampling sites (M2-M4),M2 is the subglacial soil, M3 is subglacial lake soil, M4 is soil beside glacial runoff. Water samples were directly collected into TWIRL’EM sterile sampling bags (Labplas Inc., Canada). The microbial samples were then collected by filtering 1000 mL of the water samples. The microbial biomass was successively trapped onto 47-mm-diameter, 0.2-μmpore-size membrane filters (Pall Corporation, USA). Membrane filters were placed in centrifuge tubes at −20°C in the Yellow River Station (China’s Arctic Yellow River Station is in Ny-Ålesund, Svalbard) and taken to the laboratory by plane. Filters were then frozen at −80°C until nucleic acid extraction. Surface soils sample (0–5 cm) were collected in triplicate (approximately 1 m from each other) and placed into TWIRL’EM sterile sampling bags (Labplas, Sainte-Julie, QC, Canada). Samples were stored at –80°C at China’s Arctic Yellow River Station before being transported on ice by air to the home laboratory, at which they were stored at –80°C until DNA extraction.
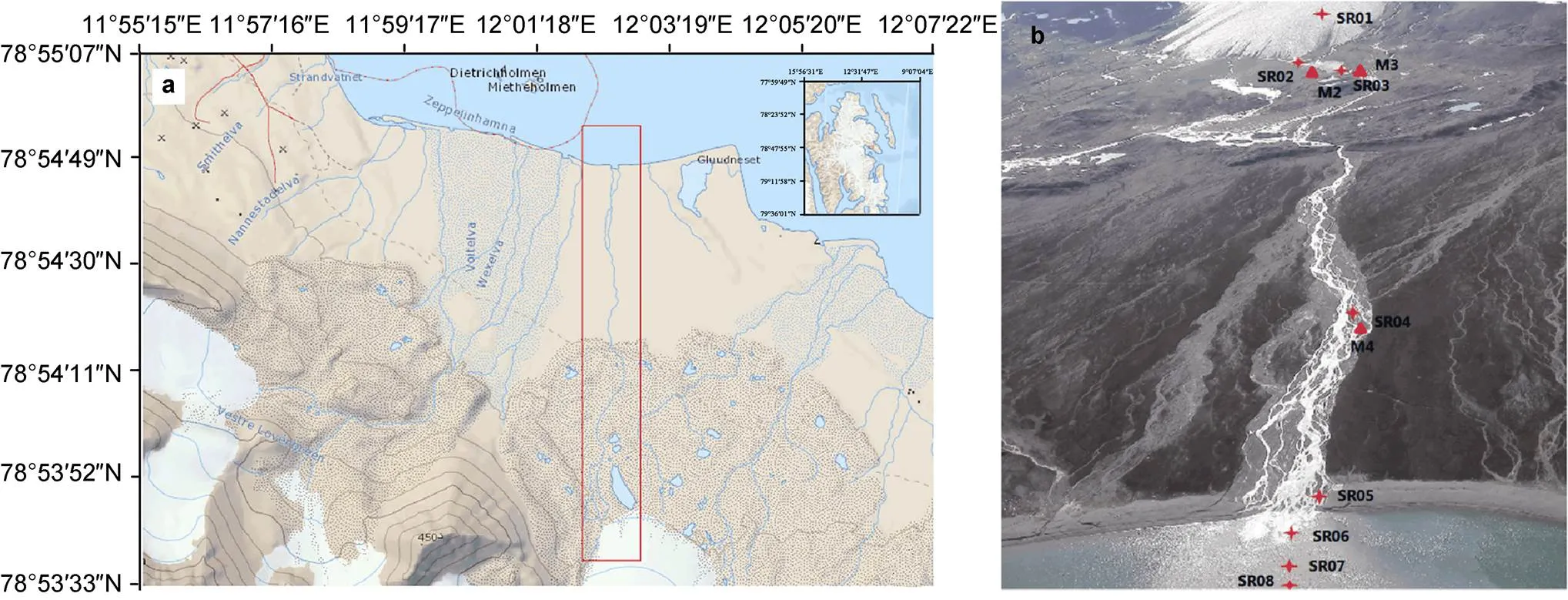
Figure 1 Location (red square; a) and image (b) of the sampling sites from Midre Lovénbreen glacier area in the present study.
2.2 Geochemical properties of the sample
A total of eight soil properties were measured, including MC (Moisture Content), pH, organic C, organic N, NH4+-N, SiO42−-Si, NO2−-N and NO3−-N. Samples were dried in an oven at 105°C to constant mass to measure MC, which was determined as proportion of water loss from the wet soil wet. Soil pH was measured by adding 10 mL of distilled water to 4 g of soil and recording pH using a pH electrode (PHS-3C, Shanghai REX Instrument Factory, Shanghai, China). Analysis of organic C and organic N was performed using an Elemental Analyzer (EA3000, Euro Vector SpA, Milan, Italy). The other five properties were analyzed using a High Performance Microflow Analyzer (QuAAtro, SEAL Analytical GmbH, Norderstedt, Germany). The water chemistry of five water samples were determined, including NH4+-N, SiO42−-Si, NO2−-N, PO43−-P and NO3−-N.
2.3 DNA extraction and PCR amplification
Genomic DNA was extracted from the membrane filters of eight water samples (1000 mL) and using a PowerWater DNA Isolation Kit (MO BIO Laboratories, Inc., USA) according to the manufacturer’s instructions. For soil samples, genomic DNA was extracted from 0.25 g of wet soil per sample (three replicates per sample)using a PowerSoil DNA Isolation Kit (MO BIO Laboratories, San Diego, CA, USA). The v3-v4 region of 16S rRNA gene was amplified using primers 338F 5’-barcode-ACTCCTACGG GAGGCAGCA-3’ and 806R 5’-barcode-GGACTACHVG GGTWTCTAAT-3’. All PCR reactions were carried out in 30 µL reactions with 15 µL of Phusion HighFidelity PCR Master Mix (New England Biolabs); 0.2 µM of forward and reverse primers, and about 10 ng template DNA. The PCR amplification cycle was: initial denaturation at 98℃ for 1 min, followed by 30 cycles of denaturation at 98℃ for 10 s, annealing at 50℃ for 30 s, and elongation at 72℃ for 30 s, with a final extension of 72℃ for 5 min. PCR products were mixed with equal volume of 1× loading buffer (containing SYB green) and loaded onto 2% agarose gel for detection. Samples with a bright main strip between 400 and 450 bp were chosen for purification with Gene JET Gel Extraction Kit (Thermo Scientific, Waltham, MA, United States).
2.4 Sequencing and data analysis
16S rRNA gene amplicons were sequenced on an Illumina MiSeq platformin the Novogene Corporation (Beijing), and 300-bp paired-end reads were generated. The raw reads were deposited into the NCBI sequence read archive (SRA) database (Accession Number: SRP115724). Clean data are obtained by trimming end bases and filtering low quality bases. QIIME (version 1.7.0) (Caporaso et al., 2010) was used for quality control and the chimeras were detected by the Gold database and UCHIME Algorithm (Edgar et al., 2011), and the chimeras sequences were removed to obtain effective data. Uparse software (Version 7.0.1001) (Edgar, 2013) was used to cluster effective data into 97% identical operational taxonomic units (OTUs). The SSU rRNA database (Quast et al., 2012) of SILVA (Wang et al., 2007) and QIIME was used to species annotate and analyze the representative sequences of OTU. Finally, all samples were normalized at the same sequence depth, for the subsequent alpha and beta diversity analysis.
Statistical analyses of the alpha-diversity of each sample via Chao1, Good’s coverage estimator, and Shannon’s index (H’) were performed. The abundance-based Bray-Curtis similarity coefficient was used to examine the dissimilarity of different samples. The relationships among the bacterial communities in the 17 samples were analyzed by hierarchical clustering analysis using the R v.3.1.1 statistical software. An analysis of similarities (ANOSIM) was performed using QIIME 1.8.0 software (Caporaso et al., 2010) to determine whether different sampling sites had significantly different bacterial communities. A linear discriminant analysis effect size (LEfSe) method (Segata et al., 2011) was used to identify the significantly different bacterial groups in different sampling types (i.e., upstream water, downstream water, upstream soil, downstream soil). The relevance of environmental factors in explaining the distribution patterns of bacterial communities in different water samples or soil samples was analyzed by Bray-Curtis distance-based redundancy analysis (db-RDA) using the R v.3.1.1 statistical software. To determine the relationship between geochemical parameters and the microbiome, network analysis is used to detect modules (OTU subnetworks) associated with geochemical factors. As described by Guidi et al. (2016), the correlations of OTUs with geochemical parameters were assessed using the sparse partial least square (sPLS) (Shen and Huang, 2008) as implemented in the R package mixOmics (Rohart et al., 2017). A global network of OTUs based on relative abundance from all samples was constructed and modules were identified based on relationship (i.e., the correlation of a gene to a module eigengene, which is a 1-dimensional vector that summarizes or is representative of the relative abundance of the nodes in a module) the degree and their correlation with geochemical factors was assessed using the R package weighted correlation network analysis (WGCNA) (Ghazalpour et al., 2006). Modules were then exported, analyzed, and visualized in Cytoscape 3.6 (Shannon et al., 2003).
3 Results
3.1 Physical and chemical properties of samples
As shown in the table, in the water sample (Table 1, Additional file 2), the maximum values of NO2–-N and PO43–-P were detected in soil water under glaciers (SR02), while the maximum values of SiO42–-Si and NO3–-N were detected at lake water under the glacier (SR03) and the maximum value of NH4+-N was detected at the glacier water (SR04). In addition, the minimum value of PO43–-P can be seen at the glacial runoff water (SR04), while the minimum values of the other four nutritive salts were seen at glacier water (SR01). In the soil samples (Table 2), the maximum values of organic N and NO3–-N were seen atsoil beside glacial runoff (M4), while the maximum values of organic carbon, NH4+-N, SiO42–-Si and NO2–-N were seen at subglacial soil (M2).
3.2 Bacterial community structure at sampling sites
A total of 273071 bacterial sequences and 1293 OTUs (at the 3% evolutionary distance) were identified in the present study. The sequence number of each sample was 16063, from which 188 to 535 OTUs were recognized. The Good’s coverage estimator of the OTUs in the samples ranged from 98.99% to 99.71% (Table 3), indicating that the sequences sufficiently covered the diversity of bacterial populations in all the samples. According to Shannon’s index analysis, the bacterial diversity of soil samples (3.84-4.57) was higher than that of water samples (3.07–4.23). The OTU rarefaction curve (Additional file 1) with all seventeen samples, indicating the species diversity of the study sites should be well represented by these samples
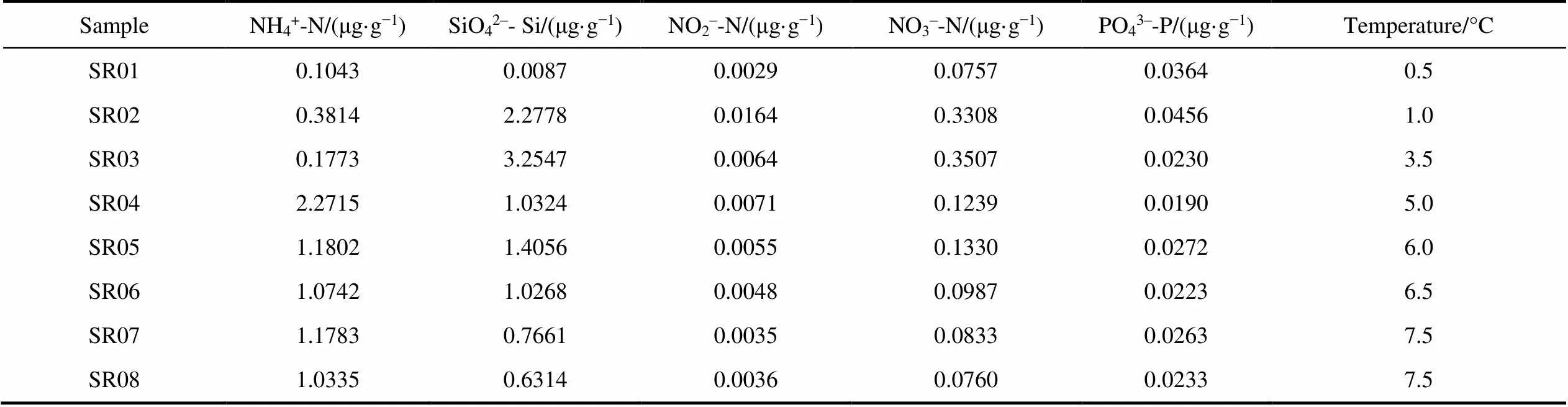
Table 1 Geochemical properties of eight water samples from glacial runoff
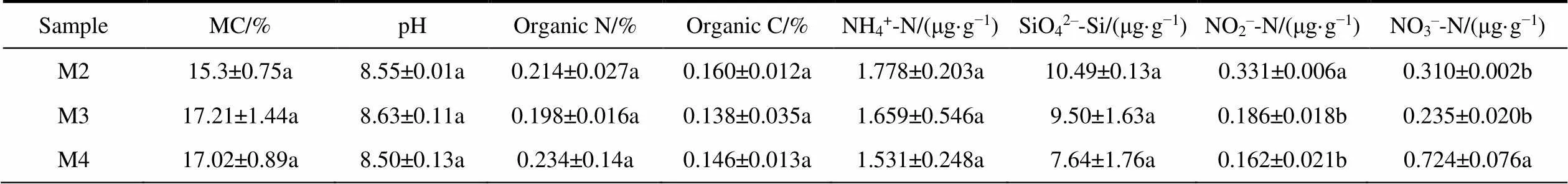
Table 2 Geochemical properties of three soil samples from glacial runoff
Notes:Statistical significance was assessed by one-way analysis of variance (ANOVA) followed by Tukey’s HSD test, and significant differences were accepted when< 0.05 between the two groups. The letters a, b and c were used to show statistically significant differences.
Proteobacteria, Bacteroidetes and Actinobacteria are common in water and soil samples of all sampling points (Figure 2).It is worth noting that the abundance of soil bacterial communities such as Actinobacteria and Acidobacteria is significantly different from that of meltwater (one-way ANOVA,< 0.05; Additional file 3). In addition, the composition of the bacterial community upstream of glacial runoff was somewhat different from that of downstream. The abundance of Actinobacteria and Firmicutes in the upstream sampling points (SR01, SR02, SR03) of glacial runoff was higher than that in the downstream sampling points (SR04, SR05, SR06, SR07, SR08) (one-way ANOVA; Additional file 4). However, the abundance of Cyanobacteria was higher in the downstream (one-way ANOVA,< 0.05; Additional file 5). At the level of classes classification, Proteobacteria in the most abundant sequence belongs to the Gammaproteobacteria, Betaproteobacteria and Alphaproteobacteria. The abundance of Betaproteobacteria (19.01%–44.31%) and Gammaproteobacteria (29.63%–63.32%) was higher in the upstream of glacial meltwater, while the abundance of Alphaproteobacteria (32.97%–38.37%) and Flavobacteriia (14.32%–21.80%) was higher in the downstream.
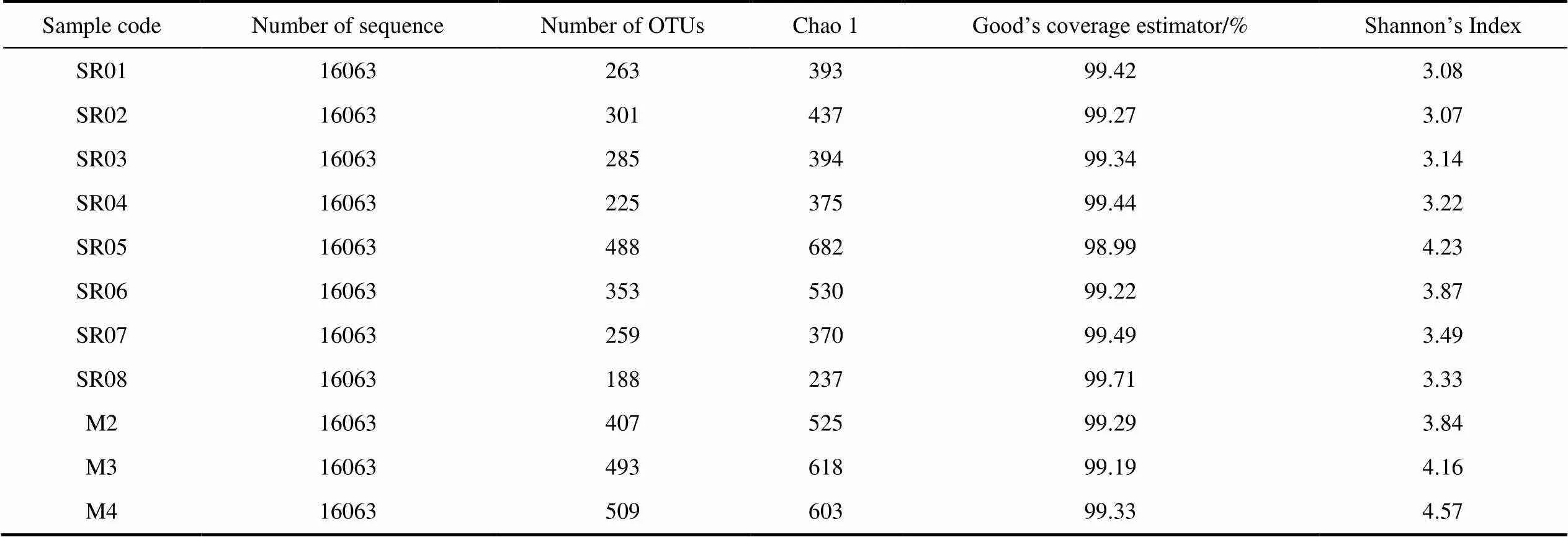
Table 3 Summary data for Miseq sequencing data from the samples in the present study
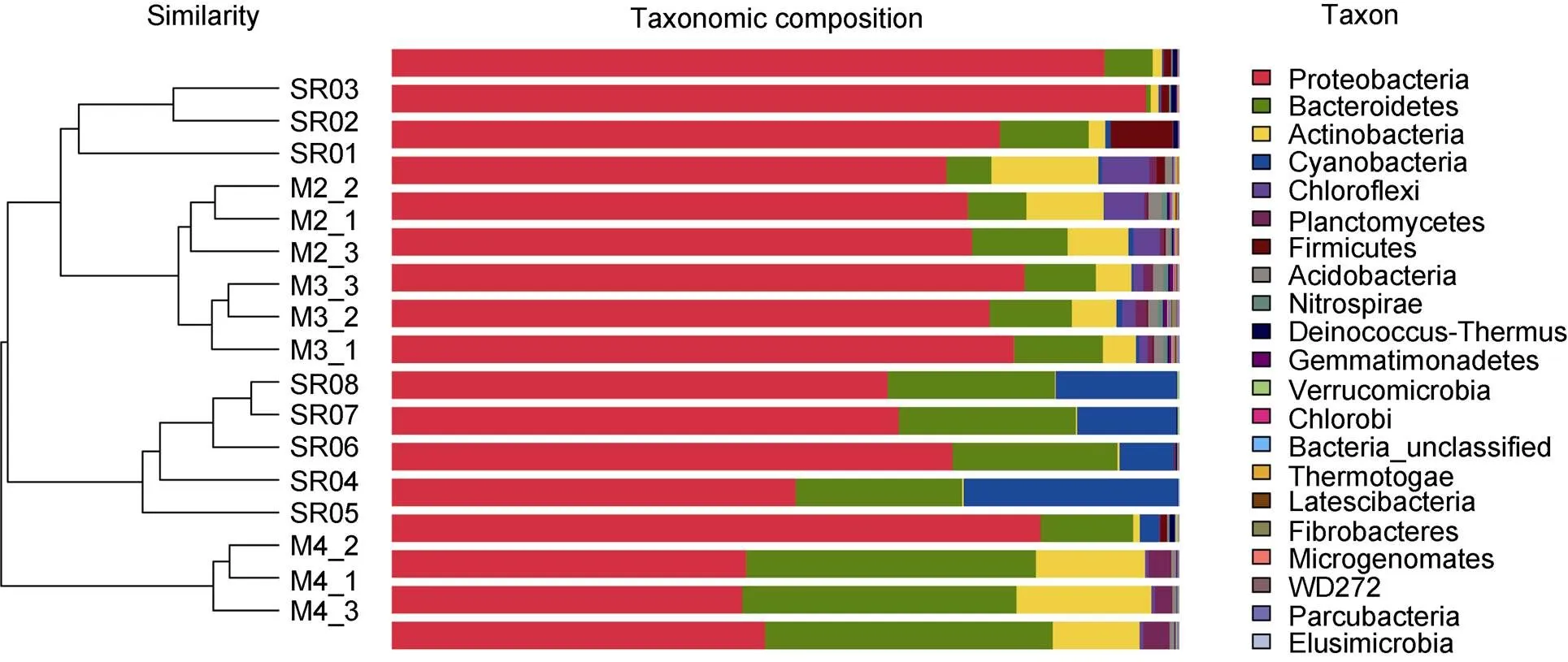
Figure 2 Bar charts of the top 21 phyla from 17 samples and cluster analysis of bacterial communities based on OTU abundance.
3.3 Bacterial differences in different sampling environments
Non-metric Multidimensional Scaling (NMDS) was used to check for differences between samples. As expected, the soil samples were separated from the water samples, and the upstream samples from the glacier runoff were isolated from the downstream samples (Figure 3). We divided the samples into four groups according to the difference, namely GroupA(SR01, SR02 and SR03), GroupB (SR04, SR05, SR06, SR07 and SR08), GroupC (M4_1, M4_2 and M4_3) and GroupD (M2_1, M2_2, M2_3, M3_1, M3_2 and M3_3). A Venn diagram demonstrated that OTUs differed among the four groups (Figure 4). The number of site-specific OTUs ranged from 20 (GroupA) to 307 (GroupD). Only 116 in 1293 OTUs were shared by all four groups.
To compare the differences in bacterial composition and structure under different environmental types, LEfSe (LDA Effect Size) analysis was conducted on four groups of sampling sites. Based on the LEfSe results, the LDA score (Linear Discriminant Analysis) of 63 taxa showed greater than 4 (the cutoff for significance test) in the 17 samples (Figure 5). GroupD has the largest number of taxa, 22 out of 63 (35%), among these groups. Enriched taxa typical in GroupD mainly includes the Bacteroidetes, Flavobacteriia, Flavobacteriaceae and Flavobacteriales. Things are different in the other groups, just like Betaproteobacteria, Burkholderiales and Comamonadaceae are enriched in GroupC and Alphaproteobacteria, Rhodobacteraceae and Rhodobacterales are in GroupB.
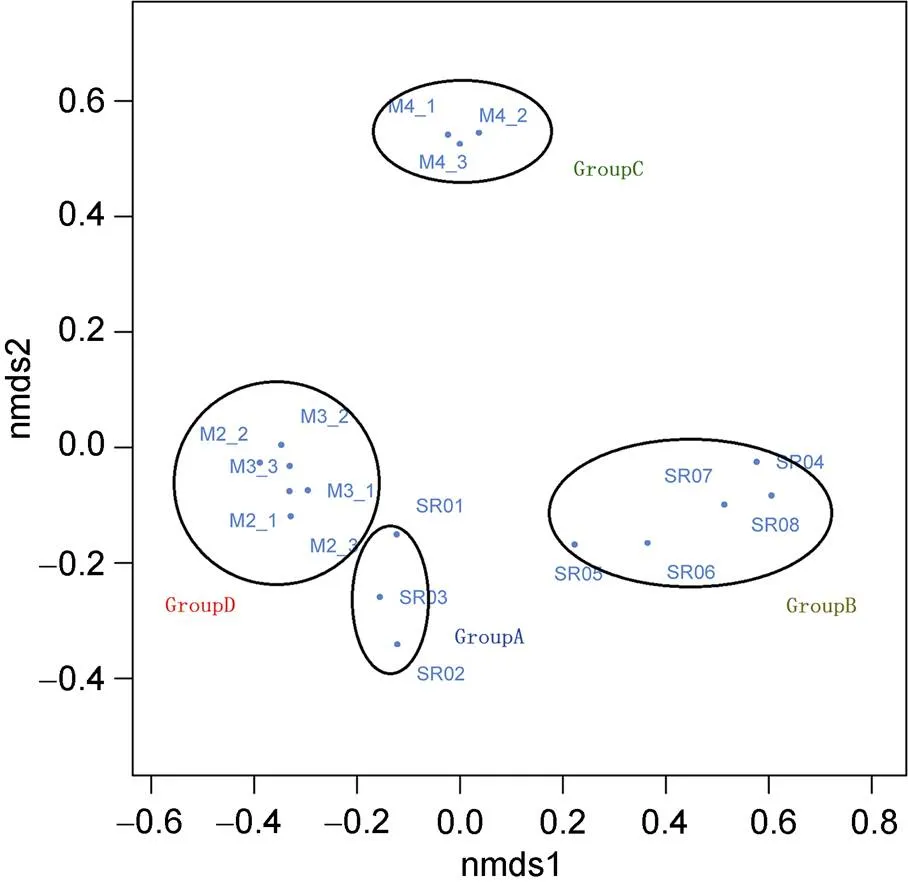
Figure 3 Non-metric Multidimensional Scaling analysis of the samples.
Figure 4 A Venn diagram displaying the degree of overlap of bacterial OTUs (at the 3% evolutionary distance) among the four groups.
3.4 Correlation between geochemical properties and bacterial community structure
The db-RDA and Monte Carlo permutation tests were performed to examine the relationship between the geochemical properties and bacterial community composition.In the RDA analysis of soil samples (RDA1 35.57%; RDA2 11.93%), the M2 samples are positively correlated with all environmental factors except pH. M3 is positively correlated with pH, SiO42–-Si and NH4+-N. M4 is positively correlated with organic N (Figure 6a). Among all the geochemical properties, NO2–-N(2= 0.9329,< 0.05) is one of the most critical factors affecting the composition of soil bacterial community (Figure 6a and Table 4). In the RDA analysis of water sample (RDA1 75.63%; RDA2 6.59%), SR01 is positively correlated with PO43–-P, SR02,SR03, SR05 and SR06 are positively correlated with NO3–-N, NO2–-N and SiO42–-Si, SR04, SR07 and SR08 are positively associated with NH4+-N (Figure 6b). Among all the geochemical properties, NO3–-N (2= 0.8196,< 0.05) is one of the most important factors affecting the bacterial community composition of water samples (Figure 6b and Table 5).
3.5 Weighted correlation network analysis of core bacteria
Weighted correlation network analysis was carried out on the OTUs with the top 300 weight values, and the expression relationship between OTUs could be shown. If the abundance of some OTUs varies in the same trend, we consider these to be in a network, thus identifying the core OTUs by its connection with others. As can be seen from the figure (Figure 7), the core OTUs of soil samples are different from those of water samples. The core OTUs of water samples are OTU983 (Sporichthyaceae), OTU599 (Cyanobacteria), OTU94 (Oceanospirillales), OTU150 (SAR11 clade, Alphaproteobacteria), OTU101 () and OTU1195 (Nitrospiraceae). while those of soil samples are OTU49 (Flavobacteriaceae), OTU53 (), OTU82 (), OTU4 (), OTU27 () and OTU42 ().
To further discuss the relationship between the core bacteria and geochemical factors and determine their correlation, we performed Pearson correlation analysis on the core OTUs obtained from network analysis and geochemical factors (Table 6 and Table 7). In water samples, OTU101 (), OTU150 (SAR11 clade, Alphaproteobacteria) and OTU1195 (Nitrospiraceae) were extremely significantly correlated with NO3–-N. In soil samples, OTU4 (), OTU27 (), OTU42 (), OTU49 (Flavobacteriaceae), OTU53 () and OTU82 () are extremely significantly correlated with NO2–-N.
4 Discussion
Glacier habitats are recognized as great repositories of microbial ecology. But most of the major taxa in glacial habitats are unculturable. In addition, the changes of bacterial community composition in the upstream and downstream of glacial runoff and the relationship between glacial runoff and soil are not fully understood. In this study, the bacterial diversity and its geochemical drivers in the runoff area of Midre Lovénbreen glacier in Svalbard were comprehensively evaluated using high-throughput sequencing technology. Arctic microorganisms are susceptible to the environmental changes of the Arctic ecosystem, and the glacial ecosystem is good sentinels of climate change in the Arctic. So understanding the Arctic microbial community is of great significance for predicting Arctic warming (Blaud et al., 2015).

Figure 5 The LDA score distribution histogram is to search for Biomarker (Segata et al., 2011), which has a statistically significant difference between group and group, at all classification levels, and used the LDA score distribution histogram to show the species with LDA score larger than 4 in the present study.
Figure 6 Distance-based redundancy analysis revealed the correlation between bacterial community and geochemical properties of soil samples (a) and water samples (b).
In the present study, the high level of Shannon diversity indices (H’=3.07–4.68) and the identification of 188–535 OTUs suggest high richness and diversity of bacterial communities in the soils samples and water samples from glacial runoff areas (Table 3). The bacterial richness observed in this study was somewhat similar to that observed in meltwater from the Southern Patagonian ice sheet (Gutiérrez et al., 2015), which the number of bacterial OTUs ranged from 47 to 589. The overall trend of OTUs increases from upstream to downstream, similar to the trend of bacterial diversity in glacial meltwater observed in Baishui Glacier (Sajjad et al., 2021).The glacier melt water systems accumulate more and more nutrients from surrounding soils, which in turn leads to the changes in microbial communities. It is of great significance to study the changes of Arctic stream biodiversity under climate warming.
Glacial meltwater flows from upstream to downstream, on the one hand, it exchanges nutrients with the nearby soil, and on the other hand, some terrestrial microorganisms enter the meltwater runoff (Hauptmann et al., 2016). These two factors result in the changes of the relative abundance of some bacterial groups upstream and downstream. Actinobacteria are common in the High Arctic, but they tend to thrive in cold, dry environmentssuch as glacial soil compared to coastal areas downstream of glacial runoff. This phenomenon can be explained by their spore formation ability, tolerance to ultraviolet radiation, and other environmental adaptations that ensure their survival in harsh conditions (Katz and Baltz, 2016; Undabarrena et al., 2016). Some genera of Actinobacteria also produce some essential bioactive compounds, including anti-fungal compounds and anti-parasitic compounds (Bérdy, 2005). The upstream of glacial runoff is closer to the glacial soil, so the abundance of Actinobacteria may be increased through the flow of soil into the glacial runoff. Some studies have shown that Firmicutes are typical Arctic tundra aerobic bacteria. For example, many studies have isolated Firmicutes from the Arctic tundra of Siberia, Svalbard and the Canadian Arctic highlands by culturable methods (Vishnivetskaya et al., 2006; Hansen et al., 2007; Steven et al., 2007). This suggests that Firmicutes are relatively common in the glacial foreland, and our results are consistent with previous ones.Several taxa in Firmicutes are endospore-forming and can cope with potentially harsh conditions (Filippidou et al., 2015). We consider Actinobacteria and Firmicutes to be typical terrigenous bacteria that enter the water through contact with glacial soil. In addition, we think Betaproteobacteria and Gammaproteobacteria are better suited to living in nutrient-rich environments (Aislabie et al., 2009). At the same time, our data show that the concentration of most nutrients is higher in upstream of glacial runoff than that in the downstream (Table 1).We analyzed that the possible reason is that the nutrient enrichment in the upstream passes through the downstream, and after reaching the estuary, the nutrient concentration decreases due to dilution.
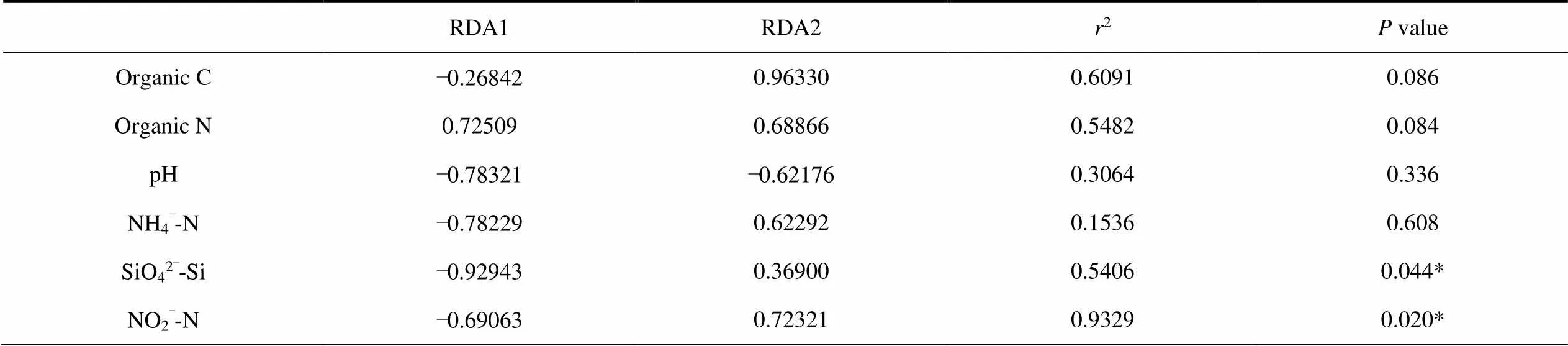
Table 4 A Monte Carlo permutation test for environmental factors and bacterial community composition of soil samples
Notes: *Correlation is significant at the 0.05 level;values based on 999 permutations.
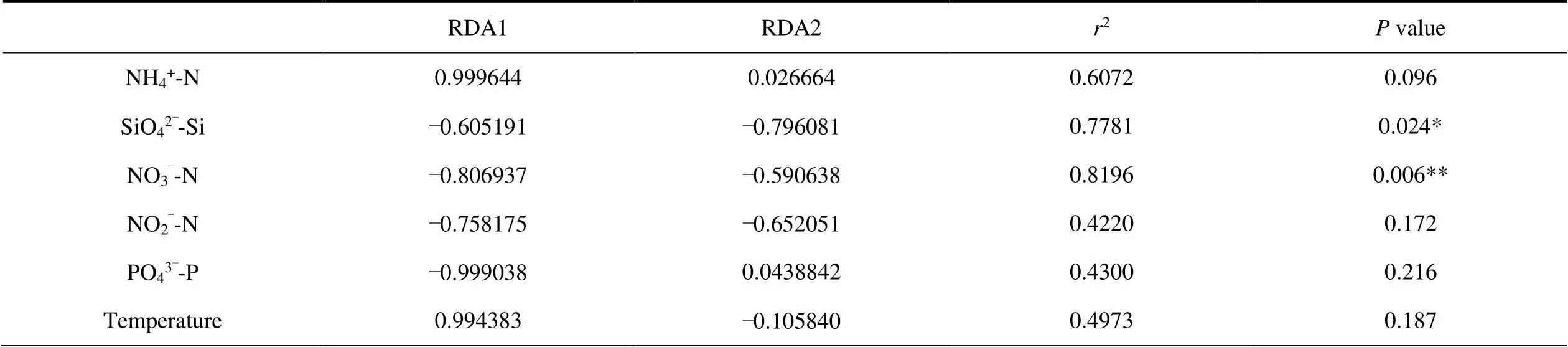
Table 5 A Monte Carlo permutation test for environmental factors and bacterial community composition of water samples
Notes: *Correlation is significant at the 0.05 level; **Correlation is significant at the 0.01 level;values based on 999 permutations.
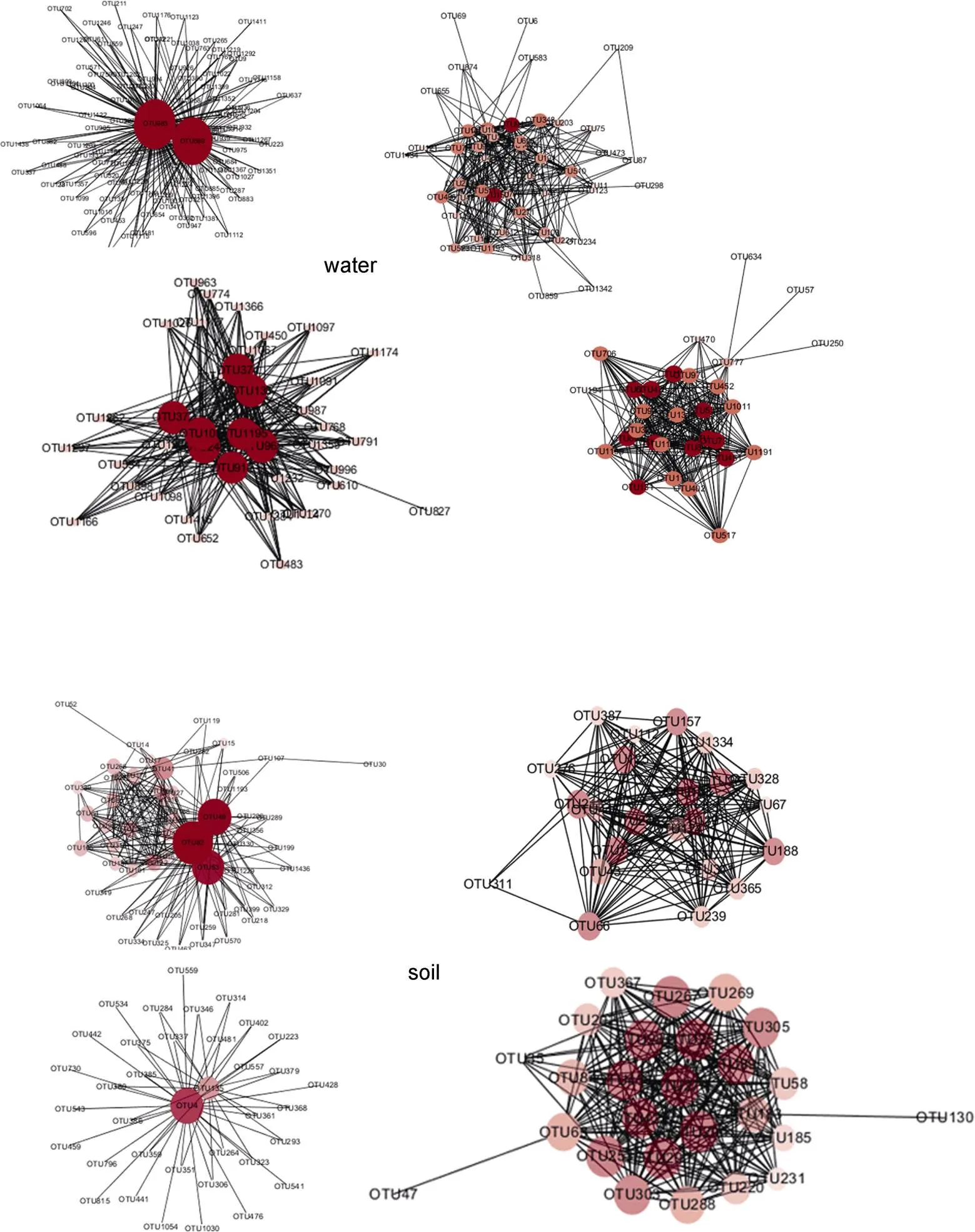
Figure 7 Weighted correlation network analysis of core bacteria in water and soil samples (The darker the color, the larger the size, and the stronger the correlation of OTU).
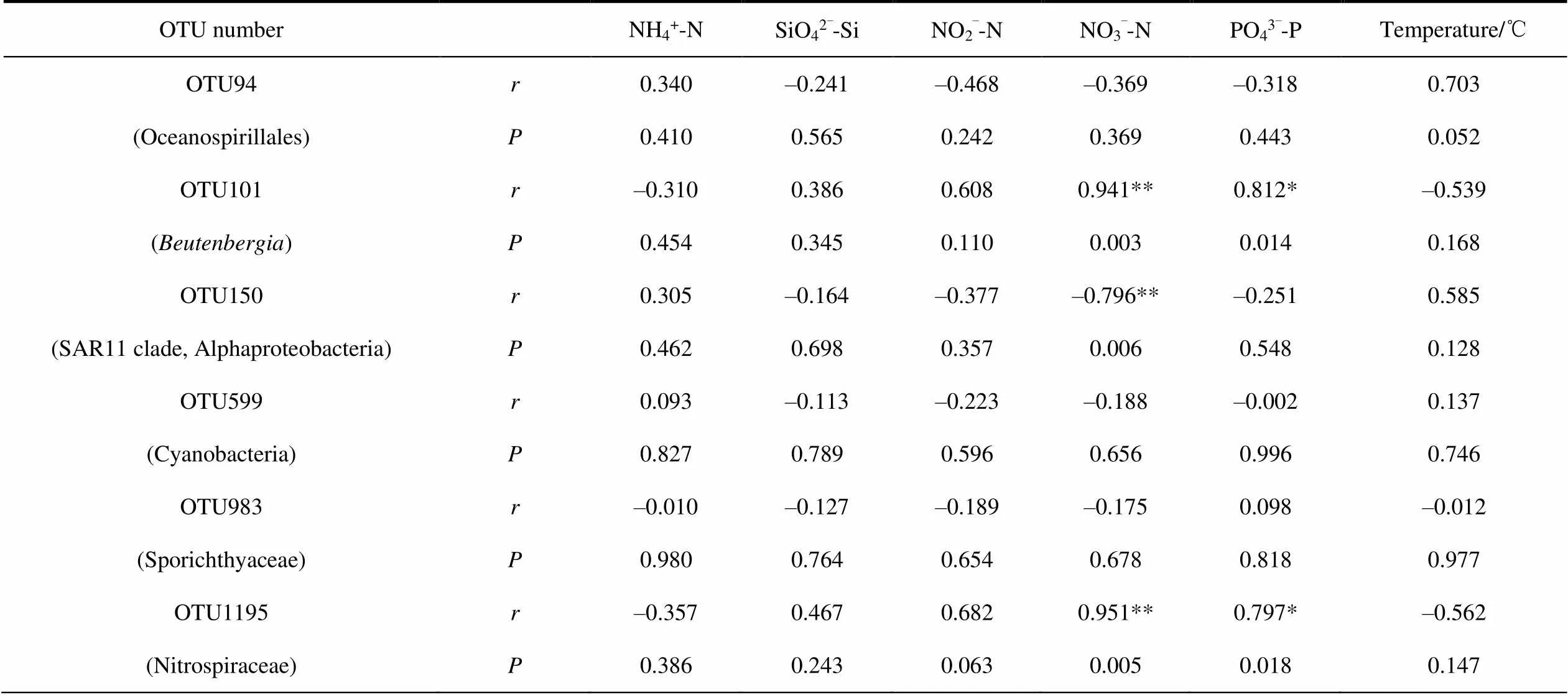
Table 6 Correlation between core OTU and geochemical factors in water samples
Notes: *Correlation is significant at the 0.05 level; **Correlation is significant at the 0.01 level.
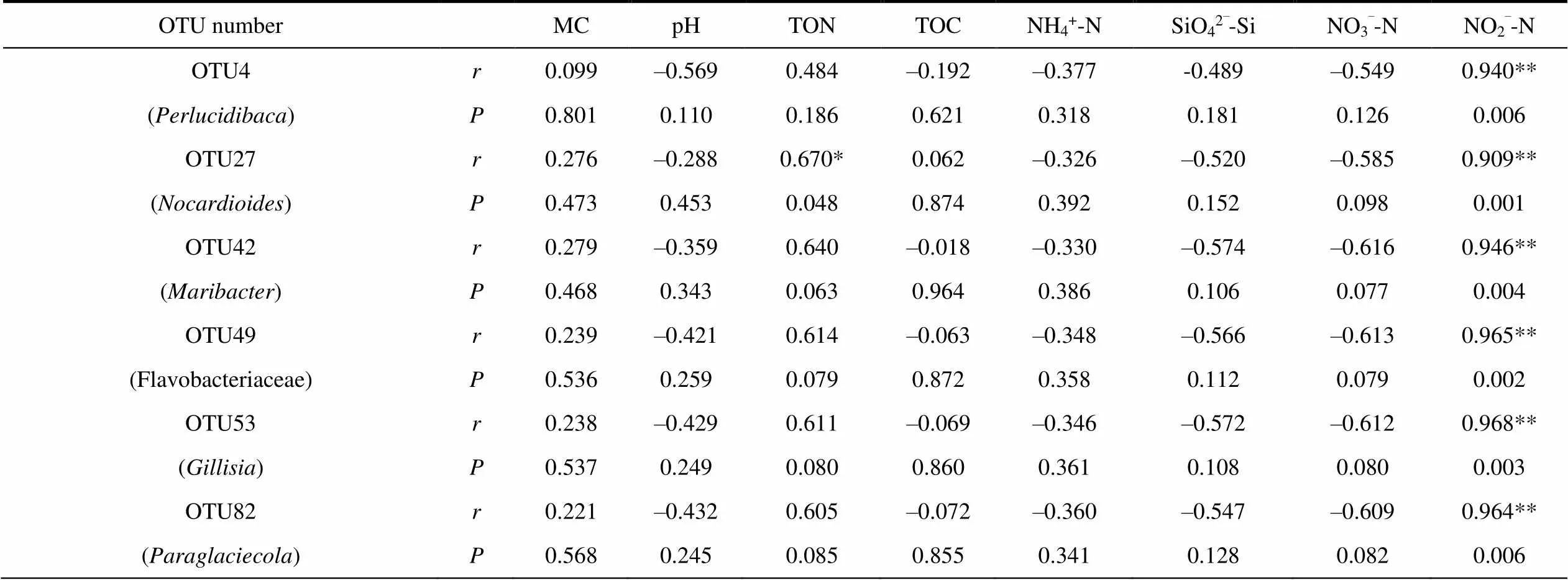
Table 7 Correlation between core OTU and geochemical factors in soil samples
Notes: *Correlation is significant at the 0.05 level; **Correlation is significant at the 0.01 level.
With climate change, sources, as well as nutritional quality, of organic matter sinking downward are expected to change in the next few decades (Wassmann and Reigstad, 2011). The intensification of glacial meltwater leads to an increase in the mass of terrestrial microbes transported into the marine environment (Lefauconnier and Hagen, 1991). This will lead to the change of microbial community structure in the downstream of meltwater runoff. Of course, the participation of some dominant microbial groups in chemical cycle as primary productivity will also change the nutrient composition of the ecosystem. Cyanobacteria, as the dominant microbial group downstream of glacial meltwater, can be involved in nitrogen fixation and conversion to ammonia, thus participating in the nitrogen cycle of marine ecosystems (Strauss et al., 2012). We believe that the higher NH4+-N in the downstream of meltwater is related to the nitrogen cycling of Cyanobacteria. In addition, the higher temperature downstream of meltwater may be responsible for the higher Cyanobacteria abundance.
Due to the ongoing interaction of meltwater runoff with adjacent soils, glacier boundaries provide an interesting ecosystem for exploring bacterial diversity. As expected, the soil samples contained high levels of NH4+-N, due to the presence of flora that enhances the soil’s nitrogen fixation. The NH4+-N content in water may be due to the periodic interaction between meltwater runoff and soil. The nutrients stored in the soil can be released toward the active layer with glacier melting (Zou et al., 2006). Beyond that, high concentrations of organic C in soil may be attributed to long-term active metabolic activities and microbial ecology. All these geochemical parameters play a crucial role in the shaping of microbial diversity in the glacial ecosystem (Yin et al., 2015).
It can be seen from the data that the OTU number and Shannon index of the soil are higher than those of the water samples (Table 3), indicating that the bacterial diversity of the glacial soil is higher than that of the glacial runoff. This could be due to several reasons, such as the fact that the soil provides a nutrient-rich solid surface for bacteria to survive while the interaction with surrounding rocks and soil increases the concentration of heavy metals in glacial runoff and reduces bacterial diversity (Sajjad et al., 2021). Moreover, higher bacterial diversity can consume soil nutrients, drive biogeochemical cycling and launch a constant ecosystem in the local niche. There is a specific relationship between the bacterial community composition of glacial soil and that of glacial runoff. For example, the abundance of Actinobacteria in glacial soils is somewhat similar to that of bacterial communities upstream from glacial runoff (Figure 2), and this may be due to the continuous interaction between water and soil and selective migration of bacteria to water (Sajjad et al., 2021). We think this is evidence of bacterial contact between glacial meltwater and soil. In addition, meltwater and soil both have their own dominant bacteria. LDA scoreanalysis showed that members belonging toandare more prominent in the soil (Figure 5). which are widely distributed in soils and have been isolated from polar and rhizosphere soils (Du et al., 2015; Chaudhary et al., 2019; Zhang et al., 2019).
Although the study area is closely related to the glacier edge, the spatial gradient of environmental factors are the main drivers of bacterial diversity and community composition. The local ecological factors act as a driving factor for the species sorting process which defines the diversity and community structure of bacteriomes (Sajjad et al., 2018). In this study, NO2–-N (<0.05) is the best predictor of soil bacterial community composition. Furthermore, organic C (< 0.05) shows a significant correlation with the bacterial community composition (Figure 6a and Table 4). In the previous studies, soil pH was found to be the most influential soil properties to determine the bacterial community structure in subarctic tundra soil (Kim et al., 2014; Shi et al., 2015), whereas Ganzert et al. (2011) reported that the soil bacterial community composition was most affected by total carbon and total nitrogen contents and soil physical factors such as moisture, but not pH. Interestingly, in present study, NO3–-N is the best predictor of bacterial community composition in the water samples (Figure 6b and Table 5).We combined WGCNA analysis to show that most of the core dominant bacteria in glacial meltwater belong to the Nitrospirae, whose members are ubiquitous nitrite oxidizers. And it have recently been reported that this kind of species is able to perform complete ammonium oxidation in a high-altitudinal and cold-water river (Liu et al., 2020).In addition, high gene abundances of nirS (nitrite reductase) and narG(nitrate reductase) have been observed in glacial foreland (Kandeler et al., 2006); on the other hand, peak nitrate reductase activity was detected in the Central Alpine glacial foreland (Deiglmayr et al., 2006).
5 Conclusion
As a dynamic ecosystem, the Midre Lovénbreen glacier meltwater has different bacterial community structures between the upstream and downstream. In addition, there are also nutrient exchange and bacterial taxa transport between the soil and the meltwater near the glacier. Our study shows that NO3–-N and NO2–-N are the most two significant geochemical factors affecting bacterial community structure in glacial meltwater and adjacent soil, respectively.
ANOVA, analysis of variance; OTU, operational taxonomic unit;RDA, redundancy analysis; WGCNA, weighted correlation network analysis;SRA, sequencing read archive; ANOSIM, analysis of similarities.
The authors declare that they have no conflict of interest.
This research was funded by the National Natural Science Foundation of China (Grant no. 41776198), the Natural Science Foundation of Shandong Province, China (Grant no. ZR2020KC036), the Key R&D Program of China (Grant no. 2018YFC1406700), and Basic Scientific Fund for National Public Research Institutes of China (Grant no. GY0219Q10). We appreciate two anonymous reviewers, and Associate Editor Prof. JonOve Hagen for their constructive comments that have further improved the manuscript.
LIN Lidong and WANG Nengfei have contributed equally to this work.
Aislabie J, Jordan S, Ayton J, et al. 2009. Bacterial diversity associated with ornithogenic soil of the Ross Sea region, Antarctica. Can J Microbiol, 55(1): 21-36, doi:10.1139/W08-126.
Ali B, Sajjad W, Ghimire P S, et al. 2021. Culture independent diversity of bacterial communities indigenous to lower altitude at Laohugou glacial environment. Geomicrobiol J, 38(1): 1-13, doi:10.1080/ 01490451.2020.1797946.
Anesio A M, Laybourn-Parry J. 2012. Glaciers and ice sheets as a biome. Trends Ecol Evol, 27(4): 219-225, doi:10.1016/j.tree.2011.09.012.
Anesio A M, Lutz S, Chrismas N A M, et al. 2017. The microbiome of glaciers and ice sheets. npj Biofilms Microbiomes, 3(10): 1-11, doi:10.1038/s41522-017-0019-0.
Bérdy J. 2005. Bioactive microbial metabolites. J Antibiot, 58(1): 1-26, doi:10.1038/ja.2005.1.
Besemer K, Singer G, Quince C, et al. 2013. Headwaters are critical reservoirs of microbial diversity for fluvial networks. Proc Biol Sci, 280(1771): 20131760, doi:10.1098/rspb.2013.1760.
Bhatia M, Sharp M, Foght J. 2006. Distinct bacterial communities exist beneath a high Arctic polythermal glacier. Appl Environ Microbiol, 72(9): 5838-5845, doi:10.1128/AEM.00595-06.
Björnsson H, Gjessing Y, Hamran S E, et al. 1996. The thermal regime of sub-polar glaciers mapped by multi-frequency radio-echo sounding. J Glaciol, 42(140): 23-32, doi:10.1017/s0022143000030495.
Blaud A, Lerch T Z, Phoenix G K, et al. 2015. Arctic soil microbial diversity in a changing world. Res Microbiol, 166(10): 796-813, doi:10.1016/j.resmic.2015.07.013.
Caporaso J G, Kuczynski J, Stombaugh J, et al. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat Methods,7(5): 335-336, doi:10.1038/nmeth.f.303.
Chaudhary D K, Dahal R H, Kim J. 2019.sp. nov., isolated from forest soil. Int J Syst Evol Microbiol, 69(9): 2762-2766, doi:10.1099/ijsem.0.003551.
Cheung S, Suzuki K, Saito H, et al. 2017. Highly heterogeneous diazotroph communities in the Kuroshio Current and the Tokara Strait, Japan. PLoS One, 12(10): e0186875, doi:10.1371/journal.pone.018 6875.
Church M J, Short C M, Jenkins B D, et al. 2005. Temporal patterns of nitrogenase gene () expression in the oligotrophic North Pacific Ocean. Appl Environ Microbiol, 71(9): 5362-5370, doi:10.1128/ AEM.71.9.5362-5370.2005.
Deiglmayr K, Philippot L, Tscherko D, et al. 2006. Microbial succession of nitrate-reducing bacteria in the rhizosphere ofacross a glacier foreland in the Central Alps. Environ Microbiol, 8(9): 1600-1612, doi:10.1111/j.1462-2920.2006.01051.x.
Du J, Singh H, Won K, et al. 2015.sp. nov., isolated from rhizosphere soil of a rose. Int J Syst Evol Microbiol, 65(Pt_9): 2949-2954, doi:10.1099/ijs.0.000361.
Edgar R C. 2013. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods, 10(10): 996-998, doi: 10.1038/ nmeth.2604.
Edgar R C, Haas B J, Clemente J C, et al. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27(16): 2194-2200, doi:10.1093/bioinformatics/btr381.
Farinotti D, Huss M, Fürst J J, et al. 2019. A consensus estimate for the ice thickness distribution of all glaciers on Earth. Nat Geosci, 12(3):168-173, doi:10.1038/s41561-019-0300-3.
Filippidou S, Junier T, Wunderlin T, et al. 2015. Under-detection of endospore-forming firmicutes in metagenomic data. Comput Struct Biotechnol J, 13: 299-306, doi:10.1016/j.csbj.2015.04.002.
Fodelianakis S, Washburne A D, Bourquin M, et al. 2022. Microdiversity characterizes prevalent phylogenetic clades in the glacier-fed stream microbiome. ISME J, 16(3): 666-675, doi:10.1038/s41396-021- 01106-6.
Ganzert L, Lipski A, Hubberten H W, et al. 2011. The impact of different soil parameters on the community structure of dominant bacteria from nine different soils located on Livingston Island, South Shetland Archipelago, Antarctica. FEMS Microbiol Ecol, 76(3): 476-491, doi:10.1111/j.1574-6941.2011.01068.x.
Ghazalpour A, Doss S, Zhang B, et al. 2006. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet, 2(8): e130, doi:10.1371/journal.pgen.0020130.
Guidi L, Chaffron S, Bittner L, et al. 2016. Plankton networks driving carbon export in the oligotrophic ocean. Nature, 532(7600): 465-470, doi:10.1038/nature16942.
Guo Y D, Wang N F, Li G Y, et al. 2018. Direct and indirect effects of penguin feces on microbiomes in Antarctic ornithogenic soils. Front Microbiol, 9: 552, doi:10.3389/fmicb.2018.00552.
Gutiérrez M H, Galand P E, Moffat C, et al. 2015. Melting glacier impacts community structure of Bacteria, Archaea and Fungi in a Chilean Patagonia fjord. Environ Microbiol, 17(10): 3882-3897, doi:10.1111/ 1462-2920.12872.
Hansen A A, Herbert R A, Mikkelsen K, et al. 2007. Viability, diversity and composition of the bacterial community in a high Arctic permafrost soil from Spitsbergen, Northern Norway. Environ Microbiol, 9(11): 2870-2884, doi:10.1111/j.1462-2920.2007.01403.x.
Hatton J E, Hendry K R, Hawkings J R, et al. 2019. Investigation of subglacial weathering under the Greenland Ice Sheet using silicon isotopes. Geochimica Cosmochimica Acta, 247: 191-206, doi:10.1016/j.gca.2018.12.033.
Hauptmann A L, Markussen T N, Stibal M, et al. 2016. Upstream freshwater and terrestrial sources are differentially reflected in the bacterial community structure along a small Arctic river and its estuary. Front Microbiol, 7: 1474, doi:10.3389/fmicb.2016.01474.
Hobbie J E, Kling G W. 2014. Alaska’s changing Arctic: ecological consequences for tundra, streams, and lakes. Oxford: Oxford University Press.
Hodson A, Anesio A M, Tranter M, et al. 2008. Glacial ecosystems. Ecol Monogr, 78(1): 41-67, doi:10.1890/07-0187.1.
Jones R T, Robeson M S, Lauber C L, et al. 2009. A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J, 3(4): 442-453, doi:10.1038/ismej.2008.127.
Junker R R, He X, Otto J C, et al. 2021. Divergent assembly processes? A comparison of the plant and soil microbiome with plant communities in a glacier forefield. FEMS Microbiol Ecol, 97(10): fiab135, doi:10.1093/femsec/fiab135.
Kandeler E, Deiglmayr K, Tscherko D, et al. 2006. Abundance of,,, andgenes of denitrifying bacteria during primary successions of a glacier foreland. Appl Environ Microbiol, 72(9): 5957-5962, doi:10.1128/AEM.00439-06.
Katz L, Baltz R H. 2016. Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol, 43(2-3): 155-176, doi:10.1007/s 10295-015-1723-5.
Kim H M, Jung J Y, Yergeau E, et al. 2014. Bacterial community structure and soil properties of a subarctic tundra soil in Council, Alaska. FEMS Microbiol Ecol, 89(2): 465-475, doi:10.1111/1574-6941.12362.
Kohler T J, Vinšová P, Falteisek L, et al. 2020. Patterns in microbial assemblages exported from the meltwater of Arctic and sub-Arctic glaciers. Front Microbiol, 11: 669, doi:10.3389/fmicb.2020.00669.
Kohler T J, Žárský J D, Yde J C, et al. 2017. Carbon dating reveals a seasonal progression in the source of particulate organic carbon exported from the Greenland Ice Sheet. Geophys Res Lett, 44(12): 6209-6217, doi:10.1002/2017GL073219.
Lefauconnier B, Hagen J O. 1991. Surging and calving glaciers in Eastern Svalbard. Norsk Polar Institutt, Medlelser MR 16, Oslo.
Li Y Y, Chen X H, Xie Z X, et al. 2018. Bacterial diversity and nitrogen utilization strategies in the upper layer of the northwestern Pacific Ocean. Front Microbiol, 9: 797, doi:10.3389/fmicb.2018.00797.
Liu S, Wang H, Chen L, et al. 2020. Comammox Nitrospira within the Yangtze River continuum: community, biogeography, and ecological drivers. ISME J, 14(10): 2488-2504, doi:10.1038/s41396-020-0701-8.
Liu Y, Yao T, Jiao N, et al. 2009. Bacterial diversity in the snow over Tibetan Plateau Glaciers. Extremophiles, 13(3): 411-423, doi:10. 1007/s00792-009-0227-5.
Luláková P, Perez-Mon C, Šantrůčková H, et al. 2019. High-alpine permafrost and active-layer soil microbiomes differ in their response to elevated temperatures. Front Microbiol, 10: 668, doi:10.3389/ fmicb.2019.00668.
Milner A M, Khamis K, Battin T J, et al. 2017. Glacier shrinkage driving global changes in downstream systems. PNAS, 114(37): 9770-9778, doi:10.1073/pnas.1619807114.
Moisander P H, Beinart R A, Hewson I, et al. 2010. Unicellular cyanobacterial distributions broaden the oceanic N2fixation domain. Science, 327(5972): 1512-1514, doi:10.1126/science.1185468.
Moisander P H, Serros T, Paerl R W, et al. 2014. Gammaproteobacterial diazotrophs and nifH gene expression in surface waters of the South Pacific Ocean. ISME J, 8(10): 1962-1973, doi:10.1038/ismej.2014.49.
Montoya J P, Holl C M, Zehr J P, et al. 2004. High rates of N2fixation by unicellular diazotrophs in the oligotrophic Pacific Ocean. Nature, 430(7003): 1027-1031, doi:10.1038/nature02824.
Noël B, Jakobs C L, van Pelt W J J, et al. 2020. Low elevation of Svalbard glaciers drives high mass loss variability. Nat Commun, 11: 4597, doi:10.1038/s41467-020-18356-1.
Pendleton S L, Miller G H, Lifton N, et al. 2019. Rapidly receding Arctic Canada glaciers revealing landscapes continuously ice-covered for more than 40, 000 years. Nat Commun, 10: 445, doi:10.1038/s41467- 019-08307-w.
Quast C, Pruesse E, Yilmaz P, et al. 2012. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res, 41(D1): D590-D596, doi:10.1093/nar/gks1219.
Vardhan Reddy P V, Shiva Nageswara Rao S S, Pratibha M S, et al. 2009. Bacterial diversity and bioprospecting for cold-active enzymes from culturable bacteria associated with sediment from a melt water stream of Midtre Lovénbreen glacier, an Arctic glacier. Res Microbiol, 160(8): 538-546, doi:10.1016/j.resmic.2009.08.008.
Rohart F, Gautier B, Singh A, et al. 2017. mixOmics: an R package for 'omics feature selection and multiple data integration. PLoS Comput Biol, 13(11): e1005752, doi:10.1371/journal.pcbi.1005752.
Sajjad W, Ali B, Bahadur A, et al. 2021. Bacterial diversity and communities structural dynamics in soil and meltwater runoff at the frontier of Baishui Glacier No.1, China. Microb Ecol, 81(2): 370-384, doi:10.1007/s00248-020-01600-y.
Sajjad W, Rafiq M, Din G, et al. 2020. Resurrection of inactive microbes and resistome present in the natural frozen world: reality or myth? Sci Total Environ, 735: 139275, doi:10.1016/j.scitotenv.2020.139275.
Sajjad W, Zheng G D, Zhang G S, et al. 2018. Diversity of prokaryotic communities indigenous to acid mine drainage and related rocks from Baiyin open-pit copper mine stope, China. Geomicrobiol J, 35(7): 580-600, doi:10.1080/01490451.2018.1430873.
Segata N, Izard J, Waldron L, et al. 2011. Metagenomic biomarker discovery and explanation. Genome Biol, 12(6): R60, doi:10.1186/gb- 2011-12-6-r60.
Shannon P, Markiel A, Ozier O, et al. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 13(11): 2498-2504, doi:10.1101/gr.1239303.
Shen H P, Huang J Z. 2008. Sparse principal component analysis via regularized low rank matrix approximation. J Multivar Anal, 99(6): 1015-1034, doi:10.1016/j.jmva.2007.06.007.
Shi Y, Xiang X J, Shen C C, et al. 2015. Vegetation-associated impacts on Arctic tundra bacterial and microeukaryotic communities. Appl Environ Microbiol, 81(2): 492-501, doi:10.1128/aem.03229-14.
Shiozaki T, Ijichi M, Kodama T, et al. 2014. Heterotrophic bacteria as major nitrogen fixers in the euphotic zone of the Indian Ocean. Glob Biogeochem Cycles, 28(10): 1096-1110, doi:10.1002/2014GB004886.
Steven B, Briggs G, McKay C P, et al. 2007. Characterization of the microbial diversity in a permafrost sample from the Canadian High Arctic using culture-dependent and culture-independent methods. FEMS Microbiol Ecol, 59(2): 513-523, doi:10.1111/j.1574-6941.2006. 00247.x.
Strauss S L, Garcia-Pichel F, Day T A. 2012. Soil microbial carbon and nitrogen transformations at a glacial foreland on Anvers Island, Antarctic Peninsula. Polar Biol, 35(10): 1459-1471, doi:10.1007/s 00300-012-1184-5.
Sułowicz S, Bondarczuk K, Ignatiuk D, et al. 2020. Microbial communities from subglacial water of naled ice bodies in the forefield of Werenskioldbreen, Svalbard. Sci Total Environ, 723: 138025, doi:10.1016/j.scitotenv.2020.138025.
Takeuchi N, Kohshima S. 2004. A snow algal community on Tyndall Glacier in the Southern Patagonia Icefield, Chile. Arct Antarct Alp Res, 36(1): 92-99, doi:10.1657/1523-0430(2004)036[0092:ASACOT] 2.0.CO;2.
Undabarrena A, Beltrametti F, Claverías F P, et al. 2016. Exploring the diversity and antimicrobial potential of marine actinobacteria from the Comau fjord in Northern Patagonia, Chile. Front Microbiol, 7: 1135, doi:10.3389/fmicb.2016.01135.
Venkatachalam S, Kannan V M, Saritha V N, et al. 2021. Bacterial diversity and community structure along the glacier foreland of Midtre Lovénbreen, Svalbard, Arctic. Ecol Indic, 126: 107704, doi:10.1016/j. ecolind.2021.107704.
Vishnivetskaya T A, Petrova M A, Urbance J, et al. 2006. Bacterial community in ancient Siberian permafrost as characterized by culture and culture-independent methods. Astrobiology, 6(3): 400-414, doi:10.1089/ast.2006.6.400.
Wang N F, Guo Y D, Li G Y, et al. 2019. Geochemical-compositional- functional changes in Arctic soil microbiomes post land submergence revealed by metagenomics. Microbes Environ, 34(2): 180-190, doi:10.1264/jsme2.ME18091.
Wang N F, Zhang T, Yang X, et al. 2016. Diversity and composition of bacterial community in soils and lake sediments from an Arctic lake area. Front Microbiol, 7: 1170, doi:10.3389/fmicb.2016.01170.
Wang N F, Zhang T, Zhang F, et al. 2015. Diversity and structure of soil bacterial communities in the Fildes Region (maritime Antarctica) as revealed by 454 pyrosequencing. Front Microbiol, 6: 1188, doi:10. 3389/fmicb.2015.01188.
Wang Q, Garrity G M, Tiedje J M, et al. 2007. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol, 73(16): 5261-5267, doi:10.1128/ AEM.00062-07.
Wassmann P, Reigstad M. 2011. Future Arctic Ocean seasonal ice zones and implications for pelagic-benthic coupling. Oceanography, 24(3): 220-231, doi:10.5670/oceanog.2011.74.
Wilhelm L, Singer G A, Fasching C, et al. 2013. Microbial biodiversity in glacier-fed streams. ISME J, 7(8): 1651-1660, doi:10.1038/ismej. 2013.44.
Yang G L, Hou S G, Le Baoge R, et al. 2016. Differences in bacterial diversity and communities between glacial snow and glacial soil on the Chongce Ice Cap, West Kunlun Mountains. Sci Reports, 6: 36548, doi:10.1038/srep36548.
Yin H, Niu J, Ren Y, et al. 2015. An integrated insight into the response of sedimentary microbial communities to heavy metal contamination. Sci Reports, 5: 14266, doi:10.1038/srep14266.
Zhang R, Zhang X Y, Sun X K, et al. 2019.sp. nov., isolated from Arctic tundra soil. Int J Syst Evol Microbiol, 69(12): 3745-3750, doi:10.1099/ijsem.0.003648.
Zhang T, Wang N F, Zhang Y Q, et al. 2016. Diversity and distribution of aquatic fungal communities in the Ny-Ålesund region, Svalbard (High Arctic): aquatic fungi in the Arctic. Microb Ecol, 71(3): 543-554, doi:10.1007/s00248-015-0689-1.
Zhang X J, Ma X J, Wang N L, et al. 2009. New subgroup of Bacteroidetes and diverse microorganisms in Tibetan Plateau glacial ice provide a biological record of environmental conditions. FEMS Microbiol Ecol, 67(1): 21-29, doi:10.1111/j.1574-6941.2008.00604.x.
Zou J W, Rogers W E, DeWalt S J, et al. 2006. The effect of Chinese tallow tree () ecotype on soil–plant system carbon and nitrogen processes. Oecologia, 150(2): 272-281, doi:10.1007/s 00442-006-0512-2.
The data set supporting the results of this article is available in the NCBI as a sequencing read archive (SRA) under accession no.SRP115724.
Supporting Information may be found in the online version of this article:
Additional file 1: Operational taxonomic unit (OTU) rarefaction curve
Additional file 2: ANOVA analysis of physical and chemical properties of glacial soils
Additional file 3: ANOVA analysis of relative abundances of Actinobacteria and Acidobacteria between glacial meltwater and soil
Additional file 4: ANOVA analysis of relative abundance of Actinobacteria and Firmicutes between upstream and downstream of glacial meltwater
Additional file 5: ANOVA analysis of relative abundance of Cyanobacteria between upstream and downstream of glacial meltwater
30 October 2021;
1 March 2022;
10 April 2022
, ORCID: 0000-0002-6070-2122, E-mail: wangnengfei@fio.org.cn
: Lin L D, Wang N F, Han W B, et al.Bacterial community diversity of meltwater runoff and soil in Midre Lovénbreen glacier in Ny-Ålesund, Arctic. Adv Polar Sci, 2022, 33(2): 167-180, doi:10.13679/j.advps.2021.0052
10.13679/j.advps.2021.0052
杂志排行

Advances in Polar Science的其它文章
- Numerical simulation of the dynamic effects of grounding icebergs on summer circulation in Prydz Bay, Antarctica
- Comparison of ship-based CTD measurement of Circumpolar Deep Water in the Amundsen Sea based on World Ocean Database
- Community structure of mesopelagic fauna and the length-weight relationships of three common fishes in the Cosmonaut Sea, Southern Ocean
- Inventory of unintentional POPs emission from anthropogenic sources in Antarctica
- Development of the International Polar Years and their benefits for China
- The evaluation of biological productivity by triple isotope composition of oxygen trapped in ice-core bubbles and dissolved in ocean:a review