基于CRISPR/Cas原理的转基因产品检测技术研究进展
2021-08-05谢宇宙付伟闫超杰王智朱鹏宇任永超张永相宁
谢宇宙, 付伟, 闫超杰, 王智, 朱鹏宇, 任永超, 张永, 相宁*
1.中国检验检疫科学研究院,北京100176;2.中华人民共和国锦州海关,辽宁锦州121013;3.贵州省黔南布依族苗族自治州检验检测院,贵州都匀558000
转基因产品检测技术是转基因贸易安全和有序进行的重要保障。转基因产品核酸分子检测技术是转基因产品检测最为直接、有效的方法。转基因产品核酸分子检测技术的不断创新与进步是转基因产品安全监管的重要支撑。目前,转基因产品核酸检测技术主要分为PCR 检测技术与环介导等温扩增(loop-mediated isothermal amplification,LAMP)技术两大类,这两类技术存在依赖仪器、操作复杂、检测时间较长等局限,一定程度上阻碍了口岸对于转基因产品的监管工作。因此,开发出无需仪器、操作简单、检测时间短、灵敏度高、特异性强的转基因产品检测技术有利于转基因产品监管工作质量的进一步提升。
基因组编辑技术是近年来迅速发展的一类遗传修饰技术,可以实现基因敲除、基因敲入和引入精细突变[1],而基因编辑技术的代表技术——CRISPR/Cas 技术,自问世以来,掀起了生物学界的一场技术革命,对生物技术发展产生了深远的影响。CRISPR/Cas 系统是细菌体内的获得性免疫防御系统,用来对抗入侵细菌的外源DNA、质粒和噬菌体[2]。CRISPR/Cas 系统除了被应用于基因编辑领域,也逐渐被应用于核酸分子检测领域,不断展现出高特异性、高灵敏度、无需仪器、快速准确等突出优势[3]。本文总结了近年来CRISPR/Cas 系统在检测领域中的研究进展,并介绍了转基因产品检测技术现状,同时对CRISPR/Cas 系统在转基因产品检测上的前景加以展望,旨在为相关人员将CRISPR/Cas 技术应用于转基因检测领域而进一步开发出适合于口岸现场快速检测的新型转基因产品核酸检测技术提供参考。
1 CRISPR/Cas检测技术研究进展
细菌体内的CRISPR/Cas 系统在对抗外源DNA、质粒和噬菌体等入侵时,首先将入侵者DNA 整合到自身CRISPR 序列中,然后将CRISPR序列转录成长 RNA 前体(pre-crispr RNA,pre-crRNA),并进一步切割成短RNA(CRISPR RNA,crRNA),最后形成导向 RNA(single-guide RNA,sgRNA)[4]。sgRNA 能够在细菌下次受到入侵时识别入侵者的核酸,并引导Cas 核酸酶切割入侵者的核酸[5]。CRISPR/Cas 系统分为两类,第1类CRISPR/Cas 系统Cas 核酸酶由多个Cas 蛋白组成,分为Ⅰ、Ⅲ和Ⅳ3 种亚型,第2 类CRISPR/Cas系统Cas 核酸酶为单个Cas 蛋白,分为Ⅱ、Ⅴ和Ⅵ3 种亚型[6]。张锋团队于2015 年发现了3 个新的2类效应蛋白(C2c1、C2c2 和 C2c3)[7],同年发现C2c2-crRNA 在匹配到靶RNA 后,除剪切匹配的靶RNA 外,还能非特异性地剪切环境中的单链RNA[3],这个发现为其他研究人员利用Cas 蛋白体外非特异性切割核酸特性进行检测提供了基础[8]。截至目前,CRISPR/Cas 系统里有三类单一效应蛋白(Cas12、Cas13 和 Cas14)被报道具有非特异性切割核酸特性,并被用于检测,加上原本的Cas9 蛋白,目前已经有4 种Cas 蛋白被用于检测领域。
1.1 基于Cas9的检测技术进展
CRISPR/Cas9 系统属于 2 类 Cas 系统Ⅱ型,能够由RNA 引导精准切割特定序列的DNA。利用CRISPR/Cas9 系统的特异性切割的特性,可以作为核酸扩增反应的“开关”,并进一步实现对靶标核酸序列的检测。Pardee 等[9]于2016 年首次将CRISPR/Cas9 系统运用到检测中,他们将等温toehold 生物传感技术与CRISPR/Cas9 结合,通过观察纸盘颜色的变化可以实现在3 h 内对寨卡病毒(Zika virus,ZIKV)美 洲 株 和 非 洲 株 的 分型(图1)。
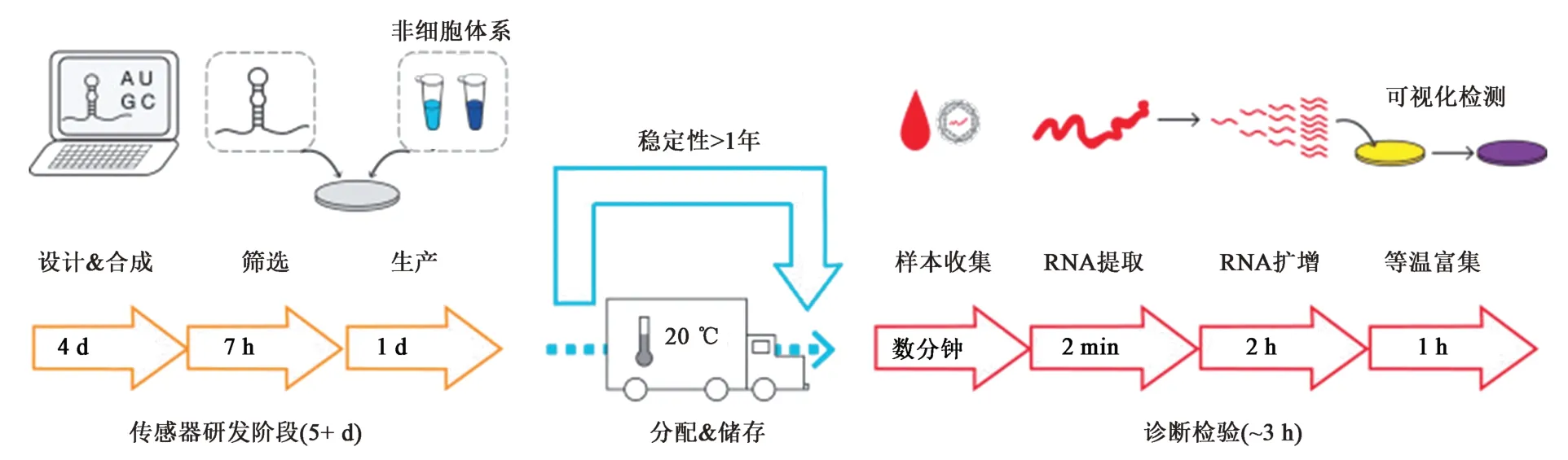
图1 CRISPR/Cas9体外分型寨卡病毒流程图[9]Fig.1 Flow chart of CRISPR/Cas9 in vitro typing of Zika virus[9]
在迈出第一步后,CRISPR/Cas9 系统与其他技术结合,被更广泛的用于检测领域。CRISDA(CRISPR-Cas9-triggered nicking endonuclease mediated strand displacement amplifi cation method)技术是CRISPR/Cas9 系统在分子检测领域的又一进步,该技术利用CRISPR/Cas系统效应蛋白Cas9在与靶核酸分子结合过程中独特的构象变化,作为链取代等温扩增反应的“开关”,高效启动针对靶核酸分子的指数倍扩增,成功检测出人基因组中乳腺癌相关的单核苷酸位点突变,并在野生型大豆中成功检测出万分之三的转基因大豆。相比于传统PCR 和其他等温扩增检测技术,该技术有以下几大优势:①能够实现aM(10-18mol·L-1)水平的高灵敏度;②达到了区分单核苷酸多态性(single nucleotide polymorphism,SNP)的特异性;③其引物设计简单,对新位点的检测反应体系开发迅速;④在90 min 内无需仪器、无需温度变化即可完成整个检测过程[10]。Huang 等[11]也将 CRISPR/Cas9基因编辑技术与等温扩增技术相结合,开发了CAS-EXPAR(CRISPR/Cas9 triggered isothermal exponential amplification reaction)技术,成功用于达到单碱基分辨率的DNA 甲基化和单核细胞增生李斯特菌总RNA 检测。除核酸检测技术外,将CRISPR/dCas9 系统和石墨烯膜场效应晶体管技术结合起来也可用于检测。以石墨烯膜为基础的晶体管上载有dCas9(只有识别功能,没有剪切功能)和sgRNA 的复合体,当样品滴加到膜上,一旦dCas9-sgRNA 识别并结合相应的靶DNA 分子,晶体管就发出一个电信号,不需要扩增样本DNA 便能够实现检测,但检测过程需要特定的仪器,无法实现现场快速检测[12]。
1.2 基于Cas13的检测技术进展
Cas13a 蛋白属于 2 类系统Ⅵ型,由 RNA 引导并切割RNA,由张锋团队于2015 年发现,当时被命名为C2c2 蛋白[7]。张锋团队于2016 年发现,C2c2/Cas13a 蛋白在crRNA 的指导下匹配到靶标RNA 后,不仅可以切割靶标RNA,还可以非特异性地剪切环境中的其他单链RNA(single-stranded RNA,ssRNA)(图 2)[8]。Doudna 团队于 2016 年首次将C2c2/Cas13a 蛋白非特异性切割RNA 特性与荧光报告RNA 结合,利用C2c2/Cas13a 蛋白在匹配到靶标RNA 后非特异性切割环境中的荧光报告RNA 分子进而释放出荧光信号,通过对荧光信号的观察实现对靶标RNA 的检测,并采用该技术成功检测了噬菌体RNA,灵敏度能够达到0.01 nM(10-9mol·L-1)[8]。
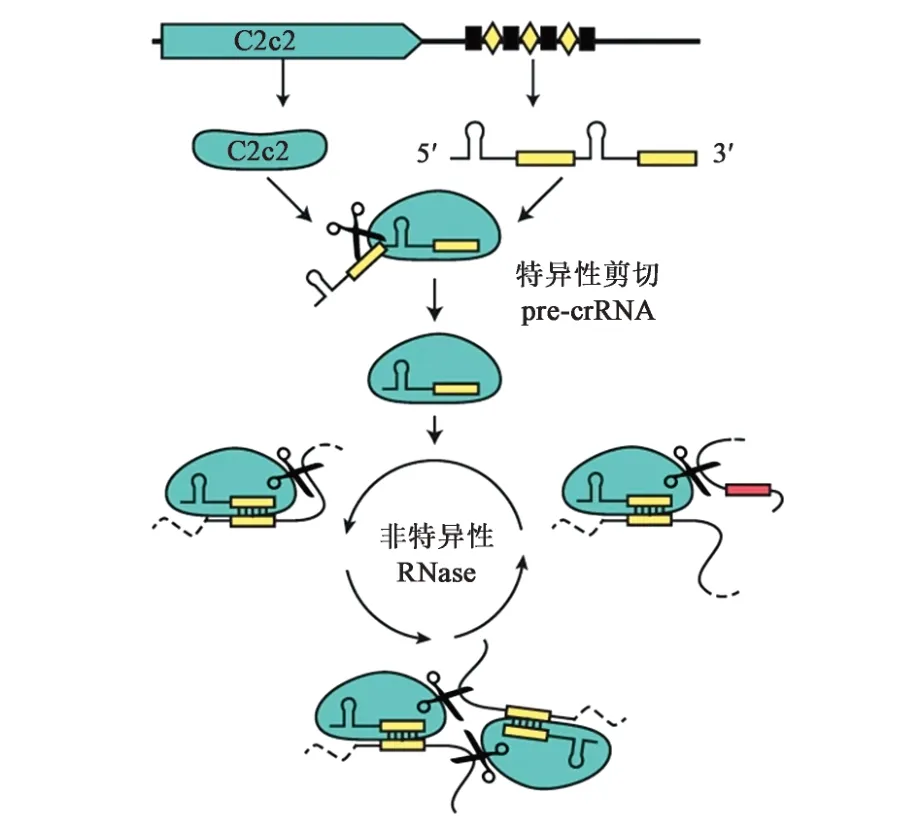
图2 Cas13a蛋白非特异性切割RNA模型[8]Fig.2 Cas13a protein non-specific cleavage of RNA model[8]
张锋团队于2017 年将Cas13a 系统非特异性切割核酸原理与重组酶聚合酶扩增(recombinase polymerase amplification,RPA)技术相结合设计了SHERLOCK(specific high-sensitivity enzymatic reporter unlocking)技术,该技术首先采用RPA 扩增DNA 或逆转录重组酶聚合酶扩增(reverse transcription-recombinase polymerase amplification,RT-RPA)技术扩增RNA,然后对DNA 产物进行转录,最后采用Cas13a-crRNA与荧光报告RNA检测转录得到的RNA(图3),并利用SHERLOCK 技术成功检测出了待测尿液样本中浓度低至每微升含单一拷贝的寨卡病毒[13]。通过引入RPA 技术,SHERLOCK 技术实现了核酸的等温扩增,同时大幅提高了灵敏度,最低能够检测到aM 水平的核酸。随后,张锋团队进一步改善了SHERLOCK 技术,在多重化方面,他们挑选了来自2个不同菌株的Cas13a 和来自2 个不同菌株的Cas13b,构建出一个单管体系拥有4 个通道的多重分析方法。在定量化方面,他们将引物系列稀释,找到了合适的引物浓度(240 nmol·L-1),改进后的 SHERLOCK技术可以实现低至2 aM 的靶标核酸定量检测。在去荧光方面,张锋团队将金标免疫层析技术与SHERLOCK 技术结合,实现了通过试纸条颜色的变化来获取结果[14]。Myhrvold 等[15]在 SHERLOCK技术的基础上设计了HUDSON(heating unextracted diagnostic samples to obliterate nucleases)技术,该技术先通过加热灭活了核酸酶与病毒,然后通过荧光显色或金标免疫层析的颜色变化实现了对患者的血清、唾液或尿样中的塞卡病毒RNA 的高灵敏度与即时检测。在新冠病毒爆发后,张锋团队设计了针对新冠病毒的检测技术STOPCovid.V2,该技术采用LAMP 技术扩增RNA,并找到了在LAMP 温度下仍能保持足够切割活性的Aap-Cas12b(Alicyclobacillus acidophilus Cas12b)酶,使得整个反应能在同一温度(60 ℃)、同一管内完成,检测下限低至100 个新冠病毒RNA 分子,灵敏度达到了93.1%,特异性达到了98.5%,整个流程只需15~45 min[16]。

图3 SHERLOCK检测原理[17]Fig.3 SHERLOCK detection principle[17]
1.3 基于Cas12的检测技术进展
Cas12a(旧称 Cpf1)属于 2 类系统Ⅴ型,由RNA 引导并负责切割单链DNA(single-stranded DNA,ssDNA),由张锋团队于 2015 年发现[18]。Doudna 团队于2018 年发现了Cas12a 非特异性切割活性,Cas12a-crRNA 复合体在识别单链或双链靶标DNA 后,能够剪切靶DNA 和非特异性地剪切环境中的ssDNA。随后,该团队将RPA 技术与Cas12a 相结合,设计了新的CRISPR/Cas 检测技术,命名为DETECTR(DNA endonuclease-targeted CRISPR trans reporter)。与 SHERLOCK 相比,DETECTR 减少了从扩增后的DNA 产物转录到RNA这一步,直接采用RPA 等温扩增待测DNA,然后将扩增产物与Cas12a-crRNA 复合体混合,识别并匹配靶标DNA,并启动Cas12a 的非特异核酸酶活性,对反应体系中的荧光报告DNA 探针进行切割,释放出荧光信号,进而实现对靶标DNA 的检测(图 4)[17]。DETECTR 技术同时能够达到 aM 水平的灵敏度和小于等于7 个碱基的特异性,Doudna 团队采用DETECTR 技术在1 h 内便准确地检测出了临床患者样本中的HPV16和HPV18并成功对其进行分型[19]。

图4 DETECTR检测原理[17]Fig.4 DETECTR detection principle[17]
与此同时,Li 等[20]也开发了与RPA 技术结合的Cas12a 检测方法,命名为HOLMES(an one-hour low-cost multipurpose highly efficient system),该方法可灵敏、特异、快速的实现SNP 分型、DNA 病毒和 RNA 病毒的检测。Li 等[21]发现 2 类Ⅴ型的Cas12b(C2c1)蛋白也具有非特异性切割能力,通过将其与LAMP 技术相结合,开发了HOLMESv2检测技术,统一了二者的反应温度,实现了快速一体化检测。Broughton等[22]将逆转录环介导等温扩增(reverse transcription loop-mediated isothermal amplification,RT-LAMP)技术、侧向流动检测技术与DETECTR 技术结合,开发出采用试纸条从呼吸拭子RNA 提取物中检测新冠病毒的方法,可在40 min内完成整个流程,特异性达到了95%,为当前基于实验室仪器的荧光定量PCR(quantitative reverse transcription PCR,qRT-PCR)新冠病毒检测方法提供了更为直观、快速的替代方法。
1.4 基于Cas14的检测技术进展
Doudna 团队在Cas12a 之后找到的新的Cas蛋白——Cas14,属于2 类系统Ⅴ型,靶向ssDNA,在激活后剪切靶ssDNA 和非特异性地剪切环境中的ssDNA。Cas14 蛋白的大小大约只有其他2类CRISPR 效应蛋白的一半,靶标序列没有前间区序列邻近基序(protospacer adjacent motif,PAM)的限制,所以可以靶向任何序列,并且对识别序列中间的碱基的要求很严格,有1 个错配就不能结合,特异性能达到单碱基。Doudna 团队仿照DETECTR 技术设计了 Cas14-DETECTR 检测技术,但由于 Cas14-sgRNA 只识别 ssDNA,因此在 RPA 扩增时,需要对引物进行修饰,降解双链DNA(double-stranded DNA,dsDNA)中的一条链,得到ssDNA。Doudna 团队通过从蓝眼和棕眼人类的唾液中提取DNA 并扩增,然后采用Cas14-DETECTR检测技术成功区分出蓝眼和棕眼的基因型,特异性达到单碱基(控制2种颜色眼睛的基因序列中只有1个碱基差异),同时实现了aM水平的灵敏度[23]。
综上所述,基于不同效应蛋白研发的CRISPR/Cas 检测技术具有不同特性,具体见表1,可在实际应用中根据需求进行选择。

表1 CRISPR/Cas检测技术比较分析Table 1 Comparative analysis of CRISPR/Cas detection technologies
2 CRISPR/Cas 检测技术在转基因产品检测上的应用进展
转基因技术是指通过体外重组DNA 技术将外源基因转入到生物体的细胞或组织中,由于导入基因的表达引起生物体性状的可遗传性修饰,从而使再生生物体获得新的遗传特性。截止到2019年,转基因作物种植面积比1996年增加了约112 倍,累计种植面积达 27 亿 hm2[24]。然而,随着转基因产品的大规模商业化,其对生态环境与人体健康的潜在风险尚存在疑问,因此需要对转基因产品进行严格的监管[25]。转基因产品检测技术是转基因产品安全评价与管理的重要技术保障。
2.1 转基因产品检测技术现状
转基因产品检测技术根据检测对象的不同主要分为两类,即针对外源核酸特定序列的检测技术与针对外源蛋白的检测技术。
针对外源核酸特定序列的检测技术是检测植物外源插入序列最直接、最有效的方法[26],可分为PCR 检测技术与LAMP 技术两大类。PCR 检测技术包括定性PCR 和定量PCR,定性PCR 中常用的有普通PCR、实时荧光定性PCR、巢式和半巢式PCR 与多重PCR[27],定量PCR 中常用的有实时荧光定量PCR 与数字PCR[28]。PCR 检测技术能够实现转基因产品的定性或定量检测,具有特异性强、灵敏度高、简便快速等优点。LAMP 技术是Notomi等[29]在2000年开发的一种新型恒温核酸扩增方法,其扩增过程不需要特殊仪器和温度循环,且兼具成本低、特异性强、灵敏度高与产物易检测等优点,适合用于现场快速核酸检测。
针对外源蛋白的检测技术是检测转基因产品中外源蛋白表达情况的直接方法,其基本原理是将表达的外源蛋白作为抗原,利用其与抗体特异性结合以实现对转基因产品快速检测,主要包括蛋白免疫印迹杂交技术(Western blot)、酶联免疫吸附检测(enzyme linked immunosorbent assay,ELISA)与免疫试纸条检测技术。相比于核酸检测技术,针对外源蛋白的检测技术可以更加直接的检测外源蛋白的表达情况,其中,免疫试纸条技术还可以作为现场初筛技术,操作简便、迅速,成本低廉[30]。
随着转基因技术的进一步商业化,转基因产品检测技术的研究与开发逐渐成为全球转基因安全监管和转基因产品国际贸易的重要支撑。尽管目前针对核酸的检测技术与针对外源蛋白的检测技术被广泛应用于转基因产品检测中,但这些技术都有自身的局限性。PCR检测技术受到反应过程中温度循环的限制,需要特定的仪器才可以完成检测,难以实现现场快速检测[31]。LAMP技术引物设计十分复杂,并且对扩增产物处理不当容易造成DNA 污染,影响后续的实验结果[32]。针对外源蛋白的检测技术,除免疫试纸条技术外,均需要依赖特定的仪器,操作复杂,同样难以实现现场快速检测。而免疫试纸条技术只能检测转基因产品中表达的外源蛋白,无法针对核酸,并且灵敏度较低,对于深加工的转基因产品(如大豆油等),因其外源蛋白含量低、抗原性差,采用免疫试纸条技术检测时的准确性较低[30]。因此,亟待开发出一种针对核酸的快速、高效、成本低廉的转基因产品现场快速检测技术,弥补当前转基因产品检测体系中现场快速检测技术的不足。
2.2 CRISPR/Cas 技术在转基因产品检测上的应用
近几年,CRISPR/Cas 技术在检测领域中迅速发展,也被逐渐应用于转基因产品检测领域。Wu等[33]利用Cas12a 系统开发了一套针对CaMV35S启动子的可视化检测方法,该方法以转基因大豆(RoundupReady®)粉中的CaMV35S 启动子作为检测靶标设计了相应的crRNA,首先通过LAMP 技术扩增靶序列,然后将CRISPR/Cas12a 荧光系统与扩增产物在37 ℃下混合5 min,在紫外光(254 nm)下就可以用肉眼清楚地识别出检测结果,成功检测到大豆粉中低至0.05%的转基因含量。Zhang等[34]结合了Cas12a系统、滤纸条基因组提取技术、RPA 技术、侧向层析检测技术设计出针对水稻叶片Cry1C基因的检测方法,该方法使用滤纸条制备基因组DNA,在体温下通过RPA 扩增目标基因,使用Cas12a 检测RPA 产物,并使用侧向层析纸条读取测试结果,可以实现无需仪器、常温、30 min 内通过观察侧向层析试纸条的颜色变化完成对转Cry1C基因水稻的鉴定。
转基因产品检测技术的不断发展是保障转基因产品贸易安全有序进行的重要支撑,新的转基因产品检测技术有利于进一步加强转基因产品的监管。传统的转基因产品现场快速检测技术主要有LAMP 技术与免疫试纸条技术,这2 种现场快速检测技术均有其局限性。CRISPR/Cas 检测技术相比于LAMP 技术,只需设计出RPA 或LAMP引物与crRNA,技术开发过程更加简单。相比于免疫试纸条技术,CRISPR/Cas 检测技术以核酸为检测靶标,拥有更高的特异性与灵敏度,针对核酸的检测也更加直接、有效。
3 展望
目前,应用在转基因产品检测上的CRISPR/Cas 检测技术主要是基于Cas12 蛋白,其特异性、灵敏度、便捷性得到了证明[33-34]。随着CRISPR/Cas 检测技术在转基因产品检测上的进一步应用,除了基于Cas12 蛋白的检测技术,Cas9 蛋白、Cas13 蛋白、Cas14 蛋白等理论上都可以设计出相应的转基因产品检测技术。但是,相比于基于Cas13蛋白的检测技术所使用的RNA 荧光报告基团,基于Cas12 蛋白的检测技术使用的是DNA 荧光报告基团,更为稳定,假阳性率较低,检测结果更加可靠。此外,基于Cas12 蛋白的检测技术的核酸扩增产物无需进行转录便可直接检测,操作更加便捷。相比于基于Cas14 蛋白的检测技术需要使用修饰的引物和T7 核酸外切酶进而生成ss-DNA 产物,基于Cas12 蛋白的检测技术来说操作更加简单。但由于CRISPR/Cas 检测技术整个检测过程都无需仪器,而只依据荧光反应或肉眼可见的颜色变化实现检测目的,因此依据荧光值强弱或颜色变化难以实现转基因产品的准确定量检测。
总体来说,CRISPR/Cas 检测技术应用于转基因产品现场快速检测时兼具以下优点:①等温扩增无需仪器,可以通过试纸条的颜色变化实现检测;②检测快速,整套检测流程可在30 min以内完成;③操作简便,普通工作者也可完成检测;④成本低廉,一套CRISPR/Cas 检测流程的成本约为4.1 美元[35];⑤设计简单,针对不同的检测序列只需重新设计RPA 或LAMP 引物和crRNA 序列;⑥高特异性,crRNA 与靶DNA 完全匹配后才能激活Cas 蛋白的非特异性切割活性;⑦高灵敏度,检测低限可达aM 水平。但目前CRISPR/Cas 检测技术在转基因产品现场快速检测中的应用仍有一些局限性,如依赖核酸扩增和富集、靶标序列受PAM 序列限制、核酸提取纯度低、Cas 蛋白的脱靶效应造成的假阳性等。但这些问题可以通过相应的改进措施来解决,如寻找Cas 蛋白的变体扩大PAM 序列识别范围[36-37]、采用高保真Cas蛋白减少假阳性[38]、采用磁性颗粒纯化DNA[39]等。
除了在转基因产品现场快速检测中应用前景广阔,CRISPR/Cas 检测技术同样在基因编辑产品的检测、转化体特异性检测以及转内源基因的转基因产品检测中展现出了巨大的潜力。在基因编辑产品的检测中,可以将基因编辑位点的特异性序列作为靶序列,设计相应的crRNA或sgRNA,若crRNA或sgRNA与特异性序列成功匹配则可启动Cas 酶的核酸切割活性,进而实现基因编辑产品的检测。在转化体特异性检测和转内源基因的转基因产品检测中,也可根据外源基因以及内参照基因的特异性序列设计相应的crRNA 或sgRNA,若crRNA 或sgRNA 与特异性序列成功匹配即可启动Cas 酶活性进而实现转基因产品的特异性检测。随着CRISPR/Cas 检测技术在转基因产品现场快速检测中的进一步应用,将会进一步促进转基因产品检测技术的发展。
