高尿酸血症模型的建立及降尿酸药物的研究进展*
2021-08-04李海涛乔攀爽
吴 芃, 王 亮, 李海涛, 乔攀爽, 张 奥, 周 虹
(北京大学医学部基础医学院药理学系,天然药物及仿生药物国家重点实验室,北京100191)
高尿酸血症(hyperuricemia)指血清尿酸(uric acid)浓度男性高于416.4 μmol/L(7.0 mg/dL),女性高于356.9 μmol/L(6.0 mg/dL)[1]。高尿酸血症的发生可能是由于尿酸生成过多和(或)肾脏、肠道排泄减少导致血清尿酸浓度过度增加[2-4]。流行病学研究显示,高尿酸血症已成为一个严重的公共卫生问题,2017 年的数据显示,我国高尿酸血症患病率为13.0%,其中男性为18.5%,女性为8.0%[5]。高尿酸血症是痛风(gout)发生发展的主要危险因素,并是多种心血管疾病和代谢性疾病的独立危险因素,包括糖尿病、高血压、动脉粥样硬化,以及慢性肾脏疾病(chronic kidney disease,CKD)[6-11]。无论是药物研发和评价还是高尿酸血症及相关疾病机制探讨都离不开高尿酸血症实验模型的建立,目前用于研究高尿酸血症的动物主要是啮齿类动物。因此,本文综述高尿酸血症小鼠、大鼠模型的构建,以及高尿酸血症细胞模型的研究与应用。
1 尿酸的代谢概述
嘌呤代谢主要在肝脏中进行,黄嘌呤氧化酶(xanthine oxidase,XO)是其限速酶,催化其最后2 个步骤,次黄嘌呤氧化产生黄嘌呤,黄嘌呤进一步氧化生成尿酸。在大多数哺乳动物中,嘌呤代谢产生的尿酸在尿酸酶(uricase/urate oxidase,UOX)的作用下进一步降解,生成溶解度更大的尿囊素。然而在灵长类动物进化过程中,编码UOX 的基因发生突变,导致人类无法将尿酸代谢为可溶性的尿囊素[12]。因此,尿酸是人类嘌呤代谢的最终产物,且人类血清尿酸水平大约是大多数其他哺乳动物的10 倍。从进化角度及其临床意义来看,人体中尿酸水平的增加具有一定的生物学功能[13],包括抗氧化和神经保护作用[14-15]、维持低盐饮食个体的血压[16]、充当固有免疫的内源性信号等[17]。
传统上,高尿酸血症被认为是嘌呤代谢障碍引起的,然而仅在少数患者中,高尿酸血症的主要原因是尿酸生成过多;在原发性高尿酸血症患者中,近端小管中尿酸重吸收增加或分泌减少占高尿酸血症发病机制的90%,越来越多的研究认为,尿酸的排泄不足在高尿酸血症的发病机理中起着核心作用[18-19]。人体内尿酸主要通过肾脏和肠道排泄,其中肾脏排泄约占2/3[20]。血浆中的尿酸在肾小球滤出,随后沿肾小管进行双向运输,该过程包括肾小管重吸收、肾小管再分泌和分泌后的再次重吸收。尿酸在肾小球的滤过分数几乎为100%,之后近端肾小管的S1段重吸收99%的尿酸,S2段再将50%的尿酸再分泌回管腔,接着S3 段再次重吸收40%的尿酸;最终尿酸的排泄分数只有10%[21]。如图1所示,肾脏对尿酸的重吸收主要由葡萄糖转运体 9(glucose transporter 9,GLUT9)和尿酸转运体1(urate transporter 1,URAT1)介导。其中,URAT1表达于近端小管上皮细胞管腔侧膜,在尿酸的重吸收中发挥主要作用[22],该蛋白也可转运促尿酸排泄药物(uricosuric drugs),如丙磺舒(probenecid)、苯溴马隆(benzbromarone)、雷西那德(lesinurad)以及最新研发的dotinurad[23],这些药物可以剂量依赖性地抑制URAT1介导的尿酸重吸收[24]。GLUT9在近端小管上皮细胞基底侧膜负责对尿酸的重吸收。钠-磷酸盐共转运体1(sodium-phosphate cotransporter type 1,NPT1)位于近端小管上皮细胞管腔侧膜上,介导尿酸的排泄。ATP 结合盒G 亚家族成员2(ATP-binding cassette subfamily G member 2,ABCG2)存在于近端小管上皮细胞和肠上皮细胞顶膜,参与肾脏和小肠的尿酸排泄,在尿酸的肾外排泄中发挥重要作用。此外,近端小管上皮细胞基底侧膜的有机阴离子转运体1(organic anion transporter 1,OAT1)和OAT3也介导尿酸分泌入管腔,负责尿酸的排泄。全基因组关联研究(Genome-Wide Association Studies,GWAS)也显示尿酸转运体在调节血清尿酸浓度的过程中起核心作用[25],其中尿酸转运体编码基因SLC2A9(编码GLUT9)、SLC22A12(编码 URAT1)、SLC17A1(编码NPT1)和ABCG2的作用最重要,血清尿酸水平主要受这4个转运体的活性调节[26]。
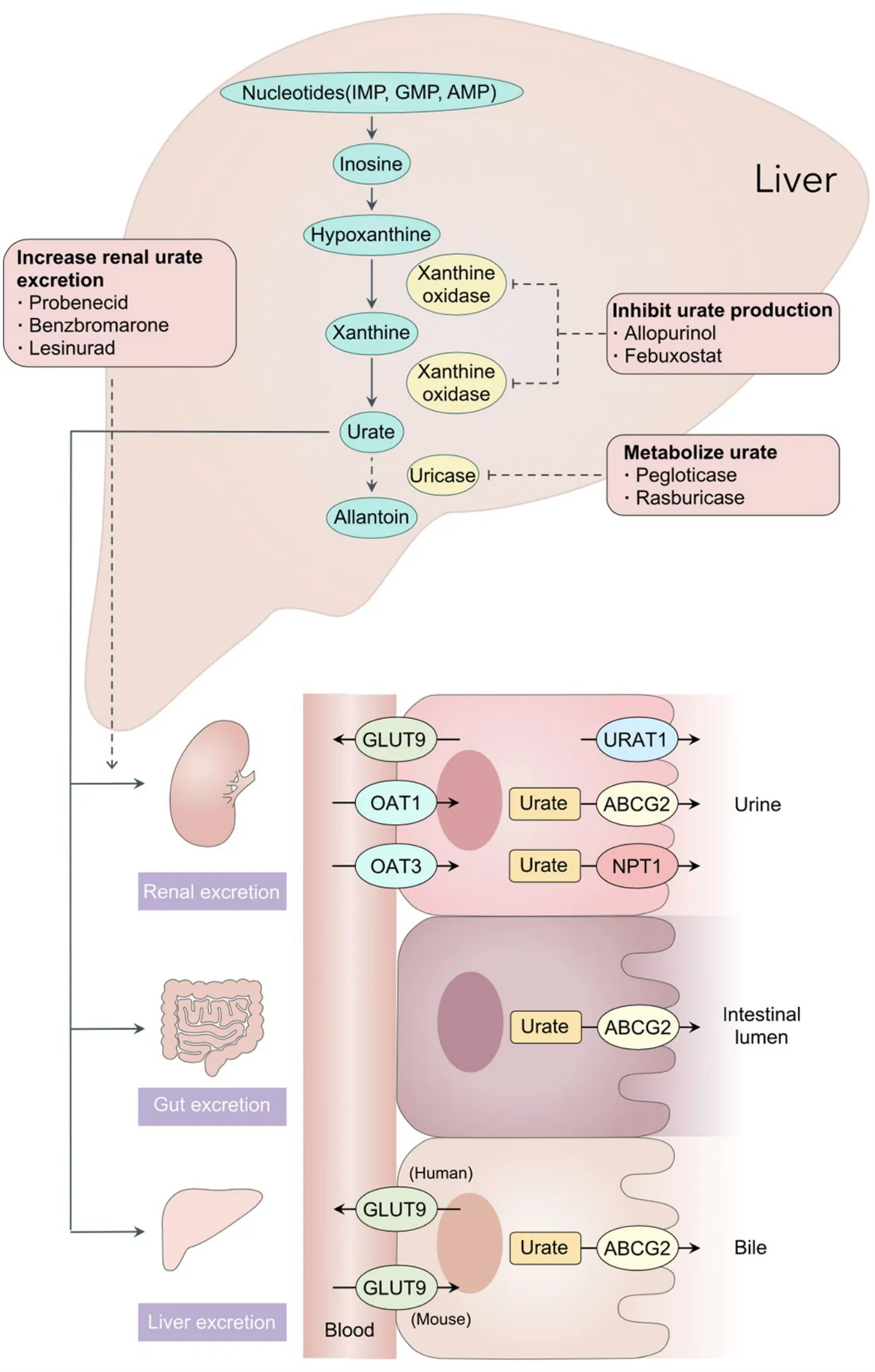
Figure 1.The production and excretion of uric acid.图1 尿酸的生成与排泄
2 高尿酸血症的治疗
目前,治疗高尿酸血症的药物主要可以分为3类:(1)抑制尿酸生成的药物,即XO 抑制剂(XO inhibitors,XOI),如别嘌呤醇(allopurinol)和非布司他(febuxostat);(2)促尿酸排泄药物,即肾尿酸转运体抑制剂,包括丙磺舒、苯溴马隆和雷西那德;(3)重组UOX,能够催化尿酸转化为水溶性更高、更容易排泄的尿囊素的药物,如聚乙二醇重组UOX 制剂普瑞凯希(pegloticase)和拉布立酶(rasburicase)[2],见图1。
根据2012 年美国风湿病学会痛风管理指南,降尿酸的一线治疗策略是使用XOI,通常是别嘌呤醇[27]。别嘌呤醇在体内迅速代谢成其活性代谢产物羟嘌呤醇,主要由肾脏排泄,随着肾功能恶化其清除率下降,易在体内蓄积,增加药物中毒风险,因此应根据肾功能调整剂量,CKD5期患者禁用[28]。相对于别嘌呤醇,非布司他对XO 的抑制效果更好、特异性更强,且主要由肝脏代谢,其清除对肾脏功能的依赖性较小[29],因此在有CKD 的情况下是更好的降尿酸药物。这2 种药物都可能引起严重的不良反应,别嘌呤醇分子中含有嘌呤,可影响嘌呤代谢的其他通路,引起诸多不良反应,表现为别嘌呤醇超敏反应综合征(allopurinol hypersensitivity syndrome,AHS),患者可出现发热、皮疹、红斑、脱屑、肾功能损伤、急性肝损伤等,病情严重可危及生命[30]。非布司他最常见的副作用是肝转氨酶升高、皮疹、恶心和关节痛,心脑血管血栓栓塞事件也有报道[31-32]。
促进尿酸排泄作为降尿酸的二线疗法,当使用XOI未达到理想血尿酸水平时,可使用丙磺舒或苯溴马隆竞争性抑制尿酸的重吸收。苯溴马隆是一种更强效的促尿酸排泄剂,通过抑制肾近端小管URAT1抑制肾小管尿酸重吸收,特别适用于肾尿酸排泄减少的高尿酸血症和痛风患者。苯溴马隆会在白种人中引起爆发性肝坏死,因此欧洲指南多推荐其作为二线药物使用[33]。但亚裔人中很少报道其肝损伤事件,这可能是因为亚裔人群的CYP2C9基因多态性不同。根据2019 年中国高尿酸血症与痛风诊疗指南,苯溴马隆被推荐为高尿酸血症与痛风降尿酸治疗的一线用药,使用过程中应密切检测肝功能[34]。雷西那德是RDEA806(一种HIV 抑制剂)的活性代谢产物,它同时抑制 URAT1 和 OAT4[35]。雷西那德于 2015 年获美国FDA批准,其早期临床试验主要是单一疗法,而后来的研究集中在与XOI联合应用,如与别嘌呤醇及非布司他联用具有进一步降尿酸的作用,但该药并未在中国上市[36-37]。2017年,美国FDA批准了首个痛风固定剂量复方药物duzallo,即200 mg雷西纳德和200或300 mg别嘌呤醇的固定剂量组合。促尿酸排泄药物的局限性包括在肾功能不全时需减少使用,而这又是在XOI难治患者中很常见的情况;另一个缺点是尿液尿酸浓度升高有诱发肾结石的风险。
重组UOX 被认为是降尿酸的三线疗法,应用较少,该方案适用于无法达到目标尿酸浓度或无法耐受其他治疗的难治性痛风患者。普瑞凯希能够迅速、大幅地降低血清尿酸水平,但20%~50%的患者发生免疫原性药物输注相关反应[38]。
许多研究和临床试验证明,XOI 和URAT1 抑制剂联合使用可降低痛风患者尿酸水平,与单一疗法相比,苯溴马隆和别嘌呤醇的组合治疗显著降低了痛风患者的血清尿酸水平[39]。因此对于单一药物充分治疗后血尿酸仍未达标的患者,可以考虑将2 种具有不同作用机制的药物联合使用以提高尿酸达标率[6]。最近,一些正在开发中的药物对XO 和URAT1均显示出双重抑制作用,其中PF-06743649(原名为KUX-1151)是第一个进入临床试验的双重抑制剂。临床研究表明,该药可导致健康受试者和痛风患者的血清尿酸水平迅速地大幅度降低。该药耐受性良好,但由于2 名受试者出现急性肾损伤而被终止进一步开发[40]。
此外,已有许多研究发现很多天然产物具有XO的抑制作用,而且对体内正常嘌呤代谢不造成影响,降尿酸效果显著,副作用小。一些天然产物还具有抗炎和抗氧化应激的作用,如槲皮素和芦丁可以阻断果糖诱导的高尿酸小鼠体内NLRP3炎症小体的激活,对肾脏起到潜在的保护作用[41-42]。在目前已有的研究中,天然产物中具有治疗高尿酸血症作用的有效成分有黄酮类(如槲皮素、芹菜素、葛根素等)、皂苷类(如穿山龙总皂苷、萆薢总皂苷和七叶莲皂苷等)、香豆素类(如秦皮总香豆素和岩白菜素等)及多酚类(如鼠尾藻多酚和茶多酚等)。这些天然产物主要通过抑制尿酸产生和促进尿酸排泄2 种机制来发挥治疗高尿酸血症作用,往往具有多环节、多层次、多靶点的调控机制,在高尿酸血症新型治疗药物的开发中具有广阔的应用前景。
3 高尿酸血症模型
由于目前已上市药物的副作用比较严重,疗效更好、副作用更小的降尿酸药物的研发至关重要,高尿酸血症模型的建立也正是研发新药的关键工具。
3.1 高尿酸血症动物模型 目前用于研究高尿酸血症的动物主要是小鼠和大鼠。高尿酸血症的动物模型可分为两大类:遗传修饰模型和化学诱导模型。遗传修饰模型主要是指通过基因修饰技术敲除UOX或尿酸转运体基因,与人类高尿酸血症的发病机制更接近;但由于UOX 和尿酸转运体是药物治疗靶点,这种模型不适合用于降尿酸药物的研究,而且其技术复杂、死亡率高,应用十分局限。化学诱导模型是利用药物增加尿酸的产生或减少其进一步降解和排泄,操作上来说更为简单,但其高尿酸水平不能长期维持,需要不断地给予造模药物,许多造模药还存在溶解度低的问题,长期给药易造成不良反应。因此,2 种模型各有利弊,需要根据研究目的和实验条件选择适当的造模动物及方法。在分析和评价大鼠和小鼠高尿酸血症模型的实验数据时,应注意的是:野生型大鼠和小鼠中的血尿酸浓度远低于人类,故尚无法明确定义其“高尿酸血症”的适当尿酸水平;并且不同研究中血清尿酸浓度的差异可能是采血方案的不同导致的,例如采血前安乐死的小鼠尿酸浓度测量值比采血前麻醉的小鼠尿酸浓度高出19 倍,采血时若未立即分离血浆或血清,则尿酸浓度会高出4 倍,这些差异是对高尿酸血症动物模型进行定量分析时需要注意的问题[43]。
3.1.1 遗传修饰模型 需要注意的是,与高尿酸血症相关的基因在人和大鼠、小鼠组织中的表达不同。例如,在人类中,编码 UOX 的基因沉默[44],而小鼠中直系同源物UOX 在多个组织中表达,并且在肝脏中高水平表达;GLUT9 主要在人肾脏中表达,而小鼠GLUT9 主要在肝脏中表达;ABCG2 主要表达于人类小肠,而在小鼠中主要在肾脏中表达[45-46]。
3.1.1.1 UOX 相关模型 由于编码UOX 的基因Uox在人类中失活,而小鼠中因为UOX的存在不能形成尿酸盐结晶,因此对小鼠Uox进行基因修饰是产生模拟人类疾病模型的重要方法。Wu等[47]报道了第一个胚胎干细胞同源性重组Uox敲除模型,通过将含有新霉素抗性基因的筛选盒(neomycin selection cassette)插入Uox的外显子3 介导Uox基因的破坏。Uox-/-小鼠的血清尿酸浓度为(654.5±101.2)μmol/L,比野生型对照组[(53.55±17.85)μmol/L]高约12倍。然而由于尿酸盐沉积,肾脏损害严重,小鼠的死亡率较高,65%的小鼠存活不超过4周,未达到性成熟,因此很难获得遗传稳定的Uox缺陷小鼠。此外,杂合交配产生Uox-/-小鼠的百分比为7.1%,远低于预期的25%,表明出现胚胎致死。Chen 等[48]发现,在饮水中加入别嘌呤醇可以适当延长Uox-/-小鼠寿命。
Lu 等[49]使用转录激活因子样效应物核酸酶(transcription activator-like effector nucleases,TALEN)技术在C57BL/6J小鼠中删除了Uox基因外显子3中28 个碱基对的区域。这些Uox-/-小鼠的尿酸水平增加,雄性为(517.7±136.9)μmol/L,雌性为(422.5±95.2)μmol/L;约有40%在出生后5 周内死亡,其死亡率远低于Wu等[47]报道的死亡率,约40%的小鼠可存活62 周。因此,该基因敲除小鼠可以获得稳定的品系,适合长期研究。杂合交配产生Uox-/-小鼠的活产率为15.9%,同样存在胚胎致死的问题。模型小鼠会发展为肾功能不全,肾脏组织出现尿酸盐沉积和肾小球或肾小管病变;雄性Uox-/-小鼠还表现出与胰岛素分泌受损相关的代谢紊乱以及对链脲佐菌素(streptozocin,STZ)诱导的糖尿病易感性增加,而雌性小鼠则发展为高血压并伴有脂代谢异常[49]。模型显示出的性别差异现象也与临床现象一致,雌激素可能是造成性别差异的潜在因素[50-52]。模型小鼠接受别嘌呤醇、苯溴马隆或非布司他治疗后,血清尿酸水平均降低,肾功能也得到改善。Uox-/-小鼠证实了高尿酸血症在代谢性疾病和高血压的发生中具有因果作用,为研究高尿酸血症及其相关的心血管系统疾病和代谢综合征提供了适宜的模型。最近Guo等[53]还利用Uox-/-小鼠模型研究了高尿酸血症对肠道的影响,证实高尿酸血症模型小鼠出现了明显的肠屏障功能障碍,肠通透性增强,可能是炎性细胞因子如肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)和白细胞介素6(interleukin-6,IL-6)的升高导致。
除了这2 个基因敲除小鼠模型外,研究人员还使用铯辐射在小鼠第3 号染色体上诱导臂内倒位[In(3)55Rk],导致小鼠Uox突变和UOX 缺乏,称之为UoxIn/In小鼠[54]。倒位纯合体小鼠的血清尿酸浓度极高:雄性为(1 255±476)μmol/L,雌性为(1 380±536)μmol/L。其中,63%的UoxIn/In小鼠在12~14日龄时死亡,而那些存活到成年的Uox缺陷小鼠通常具有正常的繁殖寿命[54]。与Uox特异性敲除小鼠相比[47,49],UoxIn/In小鼠具有更大的遗传改变,臂内倒位可能会破坏3D 核结构并对尿酸代谢有其他非特异性的影响[54]。
3.1.1.2 GLUT9 相关模型 在人类中,SLC2A9编码的尿酸转运体也称为GLUT9,主要是在肾脏中发挥尿酸重吸收作用;而在小鼠中,GLUT9主要在肝脏表达,介导肝脏对其从血液中的摄取。SLC2A9的遗传变异与人类血清尿酸水平表现出强关联性,解释了约 3% 的表型差异[25]。在 C57BL6/J 和 C57BL6/N小鼠的混合遗传背景下,利用Cre-LoxP 技术得到了全Slc2a9基因敲除模型(G9KO)和肝脏特异性Slc2a9基因敲除模型(LG9KO)[55]。与野生型小鼠(23.8~47.6 μmol/L)相比,敲除小鼠都表现出血清尿酸浓度的升高:G9KO 雄性和雌性小鼠分别为89.3 μmol/L和107.1 μmol/L,LG9KO 雄性和雌性小鼠分别为119.0 μmol/L 和 184.5 μmol/L。G9KO 小鼠患有早发性肾病,2 周龄开始出现肾脏结构明显异常。LG9KO 小鼠没有肾脏结构异常,但其血清尿酸浓度较高,这是因为GLUT9 还可以在肾脏介导对尿酸的重吸收[55]。在LG9KO 小鼠中,肌苷和高脂饮食会引起尿酸水平进一步增加,诱发慢性炎症和急性肾功能衰竭[56],但并未引起高血压[57]。该研究表明GLUT9对尿酸具有肾脏重吸收和肝脏摄取的双重功能,在尿酸稳态中发挥重要作用。G9KO小鼠中的高尿酸血症可诱发类似于人类急性高尿酸血症性肾病的肾脏疾病,因此对G9KO 小鼠的研究可能有助于研究该病的致病机制;LG9KO 小鼠的研究表明肝脏在尿酸代谢中的关键作用以及GLUT9在此过程中的作用。这些小鼠模型有助于确定血清尿酸水平长期升高在代谢综合征发展中的作用。
除了肾脏和肝脏,GLUT9 还表达于小鼠肠上皮细胞的管腔和基底膜上[58]。肠上皮细胞特异性Slc2a9基因敲除(G9EKO)小鼠的血清尿酸浓度为172.6 μmol/L,与野生型小鼠(136.9 μmol/L)相比有所增加,但差异不显著;肠道尿酸转运受损导致这些小鼠出现了代谢综合征,包括原发性高血压、血脂异常和脂肪肝[58]。别嘌呤醇治疗能够降低模型小鼠的血尿酸水平,并改善部分的代谢指标,如降低血压和总胆固醇水平。该模型小鼠生育力正常,可能适用于作为高尿酸血症相关代谢综合征的模型。
3.1.1.3 ABCG2相关模型 在人类中,ABCG2编码分泌性尿酸转运体,除肾脏外,在小肠亦发挥着尿酸分泌作用,且在肾脏排泄过饱和、肾功能不全和(或)肾脏尿酸排泄受阻时,ABCG2 介导的肠道尿酸排泄占主要地位[3,59-61]。Takada 等[62]的研究表明,Abcg2敲除(Abcg2-/-)小鼠的血清尿酸水平升高,证实了ABCG2介导的肠道尿酸排泄是重要的“肾外”尿酸排泄途径。Ichida等[3]的研究显示,当Abcg2-/-小鼠同时与氧嗪酸盐(UOX 抑制剂)联用时,与野生型小鼠(130.9 μmol/L)相比,肾脏尿酸排泄量代偿性增加而肠道尿酸排泄量不足,从而使血清尿酸浓度升高到166.6 μmol/L。
ABCG2基因单核苷酸多态性与高尿酸血症和痛风的发生密切相关,如Q140K(相当于人类中的风险变异体Q141K,SNP rs2231142)等对血尿酸水平具有重要影响[63]。用 CRISPR-Cas9 技术获得 Q140KAbcg2小鼠,其ABCG2蛋白第140位的氨基酸发生由谷氨酰胺到赖氨酸的点突变,雄性Q140K-Abcg2小鼠与野生型小鼠相比,血清尿酸浓度增加了89.4%,而雌性小鼠血清尿酸浓度无明显变化[64]。
3.1.2 化学诱导模型 相比利用基因工程技术获得具有遗传修饰的动物模型,化学诱导方法操作更为简便,药物易于获得,故目前研究大多采用此法。根据尿酸的代谢途径,可以使用药物增加尿酸来源和(或)减少去路来造模:(1)增加来源即通过补充尿酸或其前体、改变饮食等方法增加尿酸的生成;(2)减少尿酸的去路包括抑制UOX从而抑制尿酸转化为溶解度更大的尿囊素,以及抑制尿酸转运体而减少尿酸排泄。
3.1.2.1 增加尿酸的来源 有2 种方法用来增加尿酸的来源:
一是直接补充尿酸前体物质、腺嘌呤或直接补充尿酸。在体内,尿酸的代谢过程大致为腺嘌呤核糖核苷酸(AMP)-腺苷-肌苷-次黄嘌呤-黄嘌呤-尿酸,增加尿酸或其前体物质均可引起体内血尿酸水平升高。腺嘌呤在腺嘌呤磷酸核糖转移酶的作用下转变成AMP;同时腺嘌呤被大量摄入后会被XO 氧化为2,8-二羟基腺嘌呤(2,8-dihydroxyadenine,DHOA),DHOA 极难溶于水,其含量增多会沉积在肾小管导致肾脏损伤[65-66]。因此,腺嘌呤经常被用于建立慢性肾功能衰竭模型[67]。近年来研究证实,腺嘌呤也能够诱导小鼠高尿酸血症,连续28 d 给予小鼠75 mg/kg 的腺嘌呤,造模小鼠血清尿酸水平升高到(737.22±98.65)μmol/L,是对照小鼠血清尿酸水平[(307.00±56.61)μmol/L]的2.4 倍,模型组血清肌酐(serum creatinine,SCr)和血尿素氮(blood urea nitrogen,BUN)的浓度分别是(169.08±6.61)μmol/L和(42.11±4.05)mmol/L,而对照组分别是(74.83±3.53)μmol/L 和(24.85±1.17)mmol/L;同时,腺嘌呤显著增强了肝脏腺苷脱氨酶(adenosine deaminase,ADA)和XO 的活性,并且增加URAT1 的表达而促进尿酸的重吸收[68]。次黄嘌呤是尿酸生成的直接前体物质,单次腹腔注射给予小鼠次黄嘌呤250 mg/kg,1 h 后血清尿酸水平升高,但在3 和6 h 后血清尿酸水平即恢复至正常值甚至低于对照组,腹腔注射次黄嘌呤 500 和 1 000 mg/kg 后,1、3 和5h 时小鼠血尿酸水平显著高于对照组和250 mg/kg 组[69]。给予小鼠腹腔注射630 mg/kg 的次黄嘌呤,0.5 h 后血尿酸水平达到高峰[(623.76±49.96)μmol/L],4 h后降低一半,在注射后8 h 仍处于较高的水平,约为300 μmol/L,对照组小鼠血尿酸水平为196 μmol/L[70]。这类模型因UOX 的存在,常表现为尿酸一过性升高,难以达到长期、稳定的要求。
二是促进尿酸生成增多,通常通过饮食的改变诱导动物血清尿酸浓度升高,特别是在与高尿酸血症相关的其他合并症的情况下。酵母(yeast)在体内水解可以产生大量嘌呤碱和嘧啶碱,引起嘌呤代谢紊乱,XO 活性随之增加,从而促进尿酸的产生。饮食中补充果糖也可用于造模。在糖尿病情况下,高葡萄糖条件刺激肾小管上皮细胞上的钠-葡萄糖共转运体 2(sodium-glucose cotransporter 2,SGLT2)表达上调,从而使得肾小管对葡萄糖的重吸收增加。大量的葡萄糖进入细胞内,超出正常代谢所需,于是多余的葡萄糖通过多元醇途径进行代谢,产生果糖,随后果糖的代谢引起尿酸水平的升高。流行病学调查也显示饮食中富含果糖的含糖饮料会提高血清尿酸的水平[71]。果糖代谢途径的关键酶是果糖激酶,该酶无负反馈抑制,因此进入细胞的果糖会很快磷酸化,导致细胞内 ATP 耗竭,AMP 增多。ATP 耗竭可以活化嘌呤代谢酶,而AMP 是尿酸生成的上游,因此果糖的补充最后使尿酸水平升高。同时,高果糖摄入对肾脏尿酸排泄也有影响,可能与上调肾脏Glut9 蛋白水平、增加肾脏对尿酸的重吸收有关[72-73]。为了探究长期高果糖饮水对血清尿酸水平的影响,李丽玉等[74]用10%果糖喂养大鼠58 d 后,模型组血清尿酸水平升高,肾小球数目减少、毛细血管壁增厚、囊腔变窄。用10%果糖诱导了4 周的高尿酸血症模型,在模型组中除了血清尿酸水平升高外,还观察到了肾小球增生肥大、炎症细胞浸润和足细胞损伤[75]。由10%果糖诱导的模型表现为高尿酸血症、高血压和胰岛素抵抗,甚至代谢综合征,因此这种模型不适合用于研究单纯的高尿酸血症,而适用于研究高尿酸血症伴随代谢综合征的复杂疾病。
高脂饮食对小鼠血清尿酸浓度的影响也已经在一些研究中得到证实。在一项研究中,给雄性Nlrp3(NOD-,LRR- and pyrin domain-containing protein 3)基因敲除小鼠和野生型小鼠饲喂16 周对照饮食(11%能量来源于脂肪)、西方饮食(43%的能量来源于脂肪,0.15%来自胆固醇)或15%果糖水溶液。Nlrp3缺陷会破坏NLRP3炎症小体的形成,从而阻止生物活性IL-1β 的产生。野生型小鼠血浆尿酸浓度从对照饮食的0.7 mg/dL 增加到果糖饮食的1.7 mg/dL 和西方饮食的2.0 mg/dL,并且西方饮食可诱导小鼠肾脏炎症和纤维化,而在Nlrp3敲除小鼠中这些作用几乎完全减弱,提示NLRP3 炎症小体在饮食诱导的高尿酸血症和肾病中起着重要作用[76]。在一项关于高尿酸血症和非酒精性脂肪性肝病之间关系的研究中,在小鼠中补充高脂饮食8 周以诱导非酒精性脂肪性肝病,可在mRNA 和蛋白水平上调肝脏XO 的表达;在不改变UOX 表达和活性的情况下,血清尿酸浓度从 119.0 μmol/L 增加到 178.5 μmol/L[77]。在其他用甲硫氨酸缺乏和胆碱缺乏饮食诱导的非酒精性脂肪性肝病模型中,也观察到了相似的现象[78]。因此,通过饮食诱导模型可用于研究高尿酸血症与其他代谢疾病之间的联系与机制。
3.1.2.2 减少尿酸的去路 可以通过抑制尿酸进一步降解以及抑制尿酸的排泄从而减少尿酸的去路,达到升高血尿酸的目的。动物体内的UOX 是建立高尿酸血症模型的主要障碍,因此不论是在基因修饰模型还是药物诱导模型中,对UOX 的抑制都是造模的关键。氧嗪酸(oxonic acid)是抑制UOX 造模的常用药物,属于三氮杂苯类化合物,其结构与尿酸的嘌呤环相似,可竞争性抑制UOX 的活性,短时间内使尿酸水平升高,多用其钾盐。具体给药方式主要包括饲喂、饮水、灌胃和腹腔注射。饲喂法由于个体间食量不均,难以掌握给药剂量且浪费较多。腹腔注射法因氧嗪酸钾不溶于水,其混悬液难以注射,且慢性模型的建立需要每日进行给药造模,长期的腹腔注射很容易引起不良反应,例如腹腔积水和腹膜硬化。而灌胃法刺激性相对较轻,动物接受程度更好,适用于建立长期模型。用2%氧嗪酸饲喂大鼠7 周,可诱导轻度高尿酸血症,血清尿酸增加1.5~2倍,而BUN水平无明显变化,光镜检查显示肾组织结构正常,并且没有尿酸结晶沉积,免疫组化染色显示存在早期的间质纤维化[79]。这些结果表明用2%的氧嗪酸诱导高尿酸血症对肾脏的影响较小,适用于研究高尿酸血症和高血压之间的关系。连续7 d 氧嗪酸钾(250 mg/kg)灌胃给药可使小鼠血清尿酸浓度升高1.5~2.1 倍,伴随高尿酸血症性肾病[80-83]。用氧嗪酸钾诱导高尿酸血症动物模型,具有灵敏、简便、重复性好等特点,在高尿酸血症研究中应用普遍,特别在药物筛选方面是较为理想的模型。利用氧嗪酸钾构建小鼠模型,秦皮总香豆素中4 个成分秦皮甲素(esculin)、秦皮乙素(esculetin)、秦皮亭(fraxetin)和秦皮苷(fraxin)对高尿酸血症小鼠具有显著的降尿酸作用,并下调了SCr 和BUN 水平,作用机制是其对肾脏 mOAT1、mGLUT9 和 mURAT1(即小鼠的OAT1、GLUT9和URAT1)起到的调节作用,表明秦皮总香豆素对高尿酸血症和肾功能不全具有保护作用[84]。槲皮素(quercetin)可以通过调节肾有机离子转运体(renal organic ion transporters)和尿调节蛋白(uromodulin)的表达水平介导降尿酸和肾脏保护作用[85]。然而由于不同研究中,抑制剂剂量、造模时间、给药方法和测量血清尿酸水平等操作的差异,使得比较不同模型结果变得困难。值得注意的是,氧嗪酸只能抑制部分UOX 的活性,尿酸水平升高程度不高,因此通常采用长期给药或联用其他药物来建立持续稳定的高尿酸血症模型;此外,氧嗪酸钾除了抑制UOX 外,还可以抑制其他某些酶,如乳清酸磷酸核糖转移酶等,因此可能对研究存在潜在干扰。
尿酸排泄不足是大多数高尿酸血症患者发病的主要病因,抑制介导尿酸分泌的转运体可使血尿酸升高。抗结核药物乙胺丁醇可竞争性抑制近端小管中的尿酸分泌引起血清尿酸升高[86]。灌胃给予大鼠乙胺丁醇 250 mg·kg-1·d-1,联合皮下注射氧嗪酸钾200 mg·kg-1·d-1,连续 6 周后,模型组血清尿酸和 SCr水平显著升高,尿尿酸、24 h 尿酸排泄量、尿酸清除率、尿酸排泄分数等肾脏排泄尿酸水平指标均显著降低[87]。由乙胺丁醇加氧嗪酸钾诱导的高尿酸血症模型的发病机制与人类高尿酸血症是最为类似的,但是乙胺丁醇的肝、肾毒性限制了该模型的使用。值得一提的是,大量摄入腺嘌呤也会造成肾功能受损,继而影响尿酸的排泄从而使血清尿酸水平上升。
3.1.2.3 联合造模法 目前相对成熟、广泛应用的动物模型多为几种不同造模药物的联用,起到升高尿酸的协同作用,常见的是二联组合,即将尿酸前体、UOX 抑制剂或尿酸排泄抑制剂进行联用。联合造模具有迅速增加血清尿酸水平、延长维持时间和减少肾脏损害的优点。(1)次黄嘌呤联用氧嗪酸钾。用250 mg/kg 氧嗪酸钾皮下注射与150 mg/kg 次黄嘌呤腹腔注射联用,连续7 d每天给药,造模小鼠血清尿酸水平(135 μmol/L)是对照组(45 μmol/L)的3 倍。若单次给药250 mg/kg氧嗪酸钾和400 mg/kg次黄嘌呤,则可构建急性高尿酸血症小鼠模型,血清尿酸浓度高达约1 200 μmol/L。应用此模型,研究人员验证了非布司他和新设计合成的新型XOI——1-苯基咪唑-4-羧酸衍生物在体内的降尿酸作用[88]。次黄嘌呤联用氧嗪酸钾的造模方法具有尿酸水平高、持续时间长的优点,可用于长期高尿酸血症的研究。(2)腺嘌呤联用氧嗪酸钾或乙胺丁醇。腺嘌呤(100 mg/kg)和氧嗪酸钾(1 500 mg/kg)联用,每天灌胃给药连续4 周,能够建立高尿酸血症性肾病大鼠模型[89-91]。模型大鼠血清尿酸水平在第7天显著升高,在第21天达到峰值,是初始的3.2倍;SCr和BUN水平也显著升高,并可以观察到严重的肾间质纤维化和肾小球硬化[89]。腺嘌呤和氧嗪酸钾灌胃给药是建立高尿酸血症模型的有效方法,但由于腺嘌呤的肾脏毒性,应谨慎使用。腺嘌呤(150 mg/kg)和乙胺丁醇(250 mg/kg)联用也可以用于建立高尿酸血症性肾病模型,每天灌胃给药连续14 d 后,模型组小鼠血清尿酸水平、SCr、BUN 指标均显著升高,并可观察到肾组织中炎性细胞浸润、尿酸盐晶体沉积、肾小管上皮细胞肿胀[92]。因腺嘌呤和乙胺丁醇均具有肾毒性,可能对实验结果造成影响,采用这两种药物联用作为诱导动物模型的方法应用较少。有研究比较了3 种建立高尿酸血症大鼠模型的方法,包括I 组(腺嘌呤100 mg/kg 和氧嗪酸钾1 500 mg/kg)、II 组(腺嘌呤 150 mg/kg 和氧嗪酸钾 600 mg/kg)和 III 组(腺嘌呤 100 mg/kg 和乙胺丁醇 250 mg/kg),各组均每天给药,连续4周[93]。结果显示所有组的尿酸水平均呈上升趋势,最明显的是I 组;肾脏损害程度从高到低依次为III 组、II 组和I 组。与I 组相比,II 组更严重的肾脏损害表明100 mg/kg 的腺嘌呤是建立高尿酸血症模型的安全有效剂量。III组肾脏病理损伤最严重提示腺嘌呤和乙胺丁醇具有肾毒性。(3)尿酸与氧嗪酸混合饲喂。用2%氧嗪酸(25 mL/kg,灌胃)及添加0.1 mmol/L 尿酸的饮用水饲喂大鼠,2周后模型组大鼠血清尿酸水平达到对照组的3~6.6 倍,并在4 周的观察期内维持高水平;高尿酸血症大鼠体内血清尿酸水平升高后,收缩压、SCr 和BUN 显著升高,肾小球和肾小管间质发生纤维化[94]。这些现象提示该方法建立了长期稳定的高尿酸血症动物模型,且适用于研究高尿酸血症引起的高血压和肾脏损伤。5%氧嗪酸联合2.5%尿酸饲喂10 d,以及2%氧嗪酸和添加6 mg/dL 尿酸的饮用水饲喂6周,都有相似的结果[95-96]。而在饲喂2%氧嗪酸和1.5%尿酸35 d 的大鼠中还出现了尿酸晶体的沉积[97]。尿酸晶体沉积在肾小管和间质中,可引起细胞内溶酶体破裂和线粒体活性氧产生、释放,导致肾脏炎症损伤[98]。尿酸晶体在动物模型肾脏中的沉积与人类尿酸性肾病类似,因此尿酸性肾病研究的动物模型多采用此法。(4)氧嗪酸钾联用酵母。用含酵母多糖(60 g/kg)的饲料饲喂小鼠,联合250 mg/kg 氧嗪酸钾每日腹腔注射,在第3 天观察到血清尿酸升高,21 d 后肾脏病理切片表现出肾小管萎缩和间质炎症[99]。酵母(15 g/kg)灌胃和氧嗪酸钾(600 mg/kg)腹腔注射诱导大鼠高尿酸血症模型,连续造模14 d,模型组血清XO 活性升高,血清尿酸水平达到对照组的4 倍,尿β2-微球蛋白(反映肾小管损伤的指标)水平也显著升高,但 SCr 和 BUN 增加不显著[100]。每天酵母提取物(15 g/kg)灌胃2 次和每周腹腔注射1 次氧嗪酸钾(250 mg/kg),造模持续6 周,模型大鼠血清尿酸水平显著提高,肾脏形态明显变化,包括尿酸盐晶体沉积、炎性细胞浸润和肾小管间质纤维化[101]。氧嗪酸钾和酵母诱导模型的发病机制与高嘌呤饮食诱导的高尿酸血症相似,这种造模方法可以用来探究高尿酸血症治疗药物的效果以及人类高尿酸血症的发病机制。
3.2 高尿酸血症细胞模型 目前利用动物模型筛查具有降低尿酸活性的药物仍然有许多困难,存在着耗时长、可重复性差和效率低等问题。因此,需要建立高通量筛选模型,以提高降尿酸药物的筛选效率。离体细胞模型在这方面具有很好的优势,例如永生化细胞系培养出的人类细胞数量没有限制,无需分离,并具有更可靠的重现性;另外,所需的药物剂量更小,适用于大规模的筛选系统。目前,高尿酸血症体外模型的相关研究较少,因而也是研究的新方向。
Cleveland 等[102]采用 RNA 干扰降低 UOX 表达的方法,选择质粒pSilencer™4.1-CMV neo 作为载体连接shRNA 并稳定转染小鼠肝细胞系FL83B。与野生细胞系相比,Uox敲减细胞中UOX 的mRNA 含量降低66%。但由于技术困难,该研究并没有准确测量细胞内尿酸浓度。
在细胞培养液中添加尿酸前体也可以诱导尿酸生成。Adachi 等[103]在 AML12 小鼠肝细胞上建立起用于筛选具有抗高尿酸血症活性药物的分析系统,他们分别添加不同浓度的尿酸前体处理细胞一定时间,如各种嘌呤(黄嘌呤)、核苷(腺苷、肌苷和鸟苷)及核苷酸(AMP、IMP 和GMP),选用浓度有12.5、25和100 μmol/L,之后用UOX 法检测平衡盐溶液(细胞外)和细胞匀浆(细胞内)中的尿酸水平;最后选用鸟苷和肌苷各100 μmol/L 组合处理肝细胞120 min,能显著促进AML12 细胞产生尿酸,并且在该模型中,别嘌呤醇能剂量依赖性地降低肝细胞的尿酸水平。与此机制相似,用腺苷处理HK-2 细胞(人肾小管上皮细胞)并添加外源性XO,可建立高尿酸产生的细胞模型,使用HPLC检测细胞培养基中各种代谢物含量,模型组尿酸水平高约1 200 μmol/L;丙磺舒、别嘌呤醇和非布司他在该细胞模型上显示出降尿酸作用[104]。
4 总结
作为高尿酸血症机制研究、药物开发的关键工具,高尿酸血症模型的开发仍处于早期阶段。现有的高尿酸血症动物模型研究存在两个问题,一是缺乏公认的科学的评价指标,二是不能完全模仿人类高尿酸血症发病机制,80%人类高尿酸血症为多基因遗传病,其受环境和遗传因素的影响,与生活方式相关。遗传修饰模型,由于其模拟人类高尿酸血症相关基因多态性以及遗传变异,适用于高尿酸血症及相关疾病的机制研究;但成本高、死亡率高限制了其应用,开发出能够健康生存且繁衍多代的基因敲除小鼠是科研人员的首要任务。化学诱导模型没有UOX 或尿酸转运体的缺失,更适用于药物的筛选和应用。在新型药物的筛选方面,动物模型用于筛药具有周期长、重复性差的缺点,因此开发离体高通量筛药系统能够显著提高筛药效率;但离体模型只能通过初次筛选提供可能的候选化合物,仍需要利用动物模型进一步验证药效、探究机制。同时,具有多靶点的天然产物在治疗高尿酸血症上也展现出了巨大的潜力。高尿酸血症动物和细胞模型的建立是探究疾病机制、筛选新药的重要工具,目前尚无公认的最佳造模方法,因此研究人员应根据实验条件及研究目的来选择合适的模型,同时如何改良各种造模方法仍需不断探索。
