单分子表面增强拉曼散射的光谱特性及分析方法*
2021-08-04赵星郝祺2倪振华邱腾
赵星 郝祺2)† 倪振华 邱腾
1) (东南大学物理学院, 南京 211189)
2) (东南大学量子信息中心, 南京 211189)
单分子检测代表分子检测灵敏度的极限, 能够提供传统检测方式无法提供的物理信息, 在化学分析、分子动力学机理、蛋白质解析等领域具有广阔的应用前景, 具有重要的科学研究价值.具体而言, 单分子检测能对复杂体系中的分子进行识别和计数, 给出分子的分布信息; 也可以对单个分子在吸附、反应等过程中的实时衍变进行追踪, 研究分子动力学的内在机制.单分子表面增强拉曼散射是单分子检测领域最近兴起的一门新方法, 其特色在于具有特异性分子识别能力, 可以提供分子成键变化等动态信息.这种方法适用于研究分子的演化过程、分子与环境的电荷相互作用, 从而揭示分子的反应途径、分布状态、吸附方式、电荷交换等重要信息.单分子表面增强拉曼散射的概念提出较早, 但是缺乏高效的采集方法和精确的判定依据, 本文将对采集方法的优化进行梳理分析, 从非统计学和统计学两个角度对其进行讨论, 并重点对双分子分析检测法做详细介绍.另一方面, 由于单分子表面增强拉曼散射研究涉及各种交叉学科、内涵广泛, 相关研究需要对光谱背后的相关机制有深刻的理解和认识.为此, 本文基于当前的相关研究工作, 从分子漂移、光谱闪烁及展宽等特有现象入手, 分析了单分子表面增强拉曼散射的波动特征及其对应的物理机制, 并对其应用前景做了简要探讨.
1 单分子SERS的发展历程
表面增强拉曼散射(surface-enhanced Raman spectroscopy, SERS)技术是一种无损的分子检测技术, 它反映了分子键振动能量和转动能量信息,是一种能够对分子进行特异性识别的“指纹”光谱,被广泛用于定量分析、传感检测、催化、生物及能源等研究领域[1−11].通常情况下普通分子的拉曼信号强度很低, 需要通过贵金属纳米材料对其进行增强来实现高灵敏度的分子检测, 即“表面增强技术”.“表面增强技术”通常使用金、银、铜等贵金属材料, 这些材料的纳米颗粒在激发光的照射下, 会在表面产生集体电子振荡, 即表面等离激元振荡(surface plasmons resonance, SPR)模式, 该模式可以突破光的衍射极限, 将激发光转换为束缚于贵金属材料表面几个纳米空间的局域电磁场(“热点”)[12,13].这种局域电磁场强度极高, 可以将入射光能量汇聚在狭小的纳米空间之内, 从而放大附近探针分子的拉曼光谱信号, 即电磁场增强拉曼散射[14].与此同时, 贵金属材料中被激发的热电子与分子的最高占据轨道(highest occupied molecular orbital, HOMO)和最低未占据轨道(lower unoccupied molecular orbital, LOMO)之间会发生电荷转移, 导致分子的极化率增加, 形成化学增强拉曼散射[15−18].由于这两种增强方式往往同时存在和产生作用, 被统称为SERS技术.与传统的拉曼光谱相比, SERS技术能够显著增强分子的拉曼信号(高达1014甚至更大)[15], 从而实现痕量乃至单分子级别的分子检测[19,20].
单 分 子SERS (single-molecule SERS, SMSERS)是指在光斑内处于热点附近的单个探针分子所发出的拉曼光谱.与传统的拉曼光谱相比,SM-SERS不再是大量分子集合信号的平均, 而是能够提供分子的空间状态和所处环境的特异性光谱, 因此具有较窄的半高宽和更高的质量因子, 如图1所示[21].SM-SERS能够反映分子的各异性,从而获得分子的吸附状态、动力学规律及化学反应演化过程等重要信息[22], 可以在单分子水平上观察到纳米级尺度环境中单个分子的静态和动态事件, 具有重要的科学研究价值.

图1 SM-SERS与普通SERS光谱图的区别[21]Fig.1.Typical spectra of SM-SERS and traditional SERS[21].
SM-SERS在1997年首先由聂书明等[23]和Kneipp等[24]提出.他们在银胶溶液体系中分别观察到了超低浓度(10–11mol/L)罗丹明6G (Rhodamine 6G, Rh6G)分 子 和(10–14mol/L)结 晶 紫(Crystal violet, CV)分子的类SM-SERS信号.这种方法认为超低的分子浓度(亚纳摩级)实现的光谱学测量可近似地认为是单分子测量, 是早期SMSERS研究的主要方法[25−28].基于这种方法, 徐红星院士[26]对聚合的银纳米颗粒体系进行研究, 发现SM-SERS仅存在于由两个或多个金属纳米颗粒构成的体系中, 而在单个金属颗粒上难以实现,因此提出金属纳米间隙之间的超强局域电磁场是实现SM-SERS的必要条件.这类方法, 我们称之为超低浓度检测法, 使用起来相对比较简单方便,但是在对SM-SERS的认定上存在一定瑕疵, 因为分子在低浓度(单分子水平)体系中服从泊松分布,SM-SERS光谱的获取存在很大的随机性, 因此检测效率较低且难以区分单分子光谱与多分子光谱[22].针对以上问题, Le Ru等[29]在2006年提出双分子分析技术用于SM-SERS检测, 从统计学的角度有效地区分了单分子光谱与多分子光谱.这种方法现在被普遍认为是获得SM-SERS光谱的标准方法, 我们将在后文中对此进行详细介绍.
随着纳米技术和生物科技的发展, 研究人员也开始在更多的体系中进行SM-SERS研究, 例如在SPR材料表面引入带有单个染料分子的DNA用于构造“间隙”结构来实现具有高确定度的SMSERS检测[30,31], 如图2(a)所示, 或者将拉曼光谱仪与扫描探针显微镜等表征仪器结合来实现具有极高空间分辨率和单分子级别灵敏度的针尖增强拉曼散射光谱(tip-enhanced Raman spectroscopy,TERS)[32], 并用于SM-SERS检测等相关工作[33−38].在先前研究工作中TERS成像的空间分辨率大约只局限于3—15 nm[39], 这种较低分辨率不足以解析和研究单个分子的物理化学信息.2013年中国科学技术大学的董振超教授团队[33]利用扫描隧道显微镜并结合高强局域电磁场的针尖基底观察到了亚纳米级(空间分辨率为0.5 nm)单个靶分子不同振动模式的TERS成像, 如图2(b)和2(c)所示.该工作在国际上首次实现亚纳米级高分辨率单个分子的拉曼光谱成像, 使得研究单个分子在亚纳米空间中的分布状态及其物理化学信息成为可能.随后该研究团队利用这项技术在亚纳米级分别研究了相邻分子间的区分(如图2(d)所示), 以及相干分子间激子耦合的成像观察, 这些工作为研究分子间的相互作用、能量转移、分子吸附取向(如图2(e)所示)、DNA测序(如图2(f)所示)及蛋白质检测等提供了单分子水平的分析方法[34−38].

图2 (a) DNA在修饰的金@银纳米粒子壳层结构之间构造“间隙”结构用于SM-SERS检测[30]; (b)扫描隧道显微镜控制的TERS的示意图, 其中Vb是偏置电压, It是隧道电流[33]; (c)高分辨靶分子TERS光谱与图像[33]; (d)相邻分子间的TERS成像(上)及沿图中轨迹线进行十次连续TERS测量的相应光谱演化(下)[34]; (e)分子亚纳米分辨率STM图像(左)以及单个分子偶极矩模拟计算图(右)[36]; (f) DNA单链片段的TERS图像[38]Fig.2.(a) The Gap structure constructed by DNA modified gold@silver nanoparticles for SM SERS detection[30]; (b) schematic diagram of TERS controlled by scanning tunneling microscope, where Vb is the sampling bias voltage and It is the tunnel current[33];(c) high-resolution TERS spectrums and images of target molecules[33]; (d) TERS imaging between adjacent molecules (up) and the corresponding spectral evolution of ten consecutive TERS measurements along the trajectory in the left image (down)[34]; (e) molecular sub-nanometer resolution STM image (left) and single molecule dipole moment simulation calculation diagram (right)[36];(f) TERS image of DNA single-stranded fragments[38].
在上述的研究中, 除了讨论如何获取SM-SERS信号之外, 研究人员还发现SM-SERS信号随时间波动等不同于普通SERS光谱的实验结果, 并认为信号波动现象是验证SM-SERS的实验证据之一,然而在后续的研究中发现在多分子或其他体系中也存在该现象[22,40].虽然光谱信号波动不是一种严格证明SM-SERS的方法, 但是SM-SERS中的光谱波动却能够提供分子细微的SERS光谱, 并揭示出分子在“热点”区域的分布状态及在衬底表面的吸附方式等微观系统中的行为特征, 具有重要的研究意义.当前SM-SERS的发展仍处于初级阶段,如何获取可靠稳定的SM-SERS光谱, 以及理解SMSERS光谱波动背后的理论机制是目前研究的核心方向.基于此, 本文将针对用统计学和非统计学方法获取SM-SERS的技术进行介绍, 并对SM-SERS光谱的特性及可能的理论机理进行阐述和分析.最后, 本文就目前的研究工作提出了SM-SERS面临的挑战及未来努力的方向, 希望能为今后SMSERS研究工作的开展起到一个抛砖引玉的作用.
2 单分子SERS光谱的采集
上文已经提到, 使用超低浓度检测法的非统计学方法来获取单分子光谱事件是获取SM-SERS信号一种常见且简易的方法.该方法一方面稀释靶分子浓度以保证在平均意义上的探测体积内存在一个甚至更少的分子, 另一方面需要优化基底的结构、形貌, 以及选择合适的激发波长来实现高增强因子(enhancement factor, EF)的SERS增强.聂书明等[23]和Kneipp等[24]认为较低的分子浓度和1014以上的增强因子(enhancement factor, EF)是获得SM-SERS信号的关键.一般认为, SERS的EF来源于电磁增强和化学增强两部分[41].但除了上述两种主要的增强机制外, 分子与基底接触后, 与自由态分子相比, 其轨道能级发生杂化.当激发光的光子能量与分子HOMO和LOMO轨道之间的能级差匹配时, 形成共振增强拉曼散射(resonance-enhanced Raman spectroscopy, RERS).该现象也是SERS中必须要考虑到的增强因素, 特别是在SM-SERS研究中, 这种增强的贡献是不容忽略的.虽然RERS只能提供大约102—106倍的增强效果[22], 并且从理论上来说其较低的EF很难单独用于采集SM-SERS信号, 但是它与电磁场和化学增强的协同作用有助进一步提高靶分子的拉曼信号, 从而使SM-SERS信号采集更加稳健可靠.基于此, Habuchi等[42]利用488 nm激光在银胶体系中观察到了单分子级别绿色荧光蛋白分子的拉曼信号.除了局域电磁场的贡献之外, 作者认为RERS也有助于SM-SERS检测.在后续的研究中Blackie等[43]发现不同散射截面探针分子实现SM-SERS需要的EF有较大区别.对于散射截面比较大的共振探针分子(10–27—10–28cm2/sr), 考虑到化学增强和RERS的贡献, “热点”处的电磁场增强EF需达到108以上, 如图3所示; 对于散k射截面比较小的非共振分子(10–29—10–30cm2/sr)而言, 电磁增强EF至少在1010以上才能获得SMSERS信号[43].

图3 不同散射截面分子实现SM-SERS检测所要满足的电磁增强EF[43]Fig.3.Electromagnetic enhancement EF value of molecules with different cross sections required for SM-SERS detection[43].
这种使用超低浓度检测方法被广泛运用于早期SM-SERS的采集以及研究催化反应动力学中反应物分子之间的相互作用及化学反应的本质.例如: Zhang等[44]利用金纳米颗粒二聚体观察到了4-硝基苯硫酚(4-Nitrothiophenol, 4-NTP)的光裂解现象, 揭示了硫酚分子的生成反应途径, 如图4(a)所示, 为后续研究单分子4-NTP催化反应动力学提供了理论依据和实验支撑; 厦门大学李剑峰团队[45]在纳米腔中研究了单个罗丹明B异硫氰酸酯分子的催化反应过程, 如图4(b)所示, 该研究利用光谱闪烁等SM-SERS特性将SERS信号与分子的荧光联系起来, 揭示了纳米甚至亚纳米级别的微观系统中单个分子的化学反应行为与宏观反应的区别.

图4 SM-SERS技术用于监测原位催化反应过程 (a) 单个4-NTP分子通过原位催化反应生成硫酚分子的SERS光谱图[44];(b)单分子罗丹明B异硫氰酸酯经历不同光照时间催化生成不同物质所对应的光谱图[45]Fig.4.Monitoring in situ catalytic reaction via SM-SERS: (a) SERS spectra of Thiophenol generated by single-molecule 4-NTP via in situ catalytic reaction[44]; (b) SERS Spectra of Rhodamine B isothiocyanate catalyzed by single-molecule rhodamine B under different illumination time[45].
上述工作大多近似地认为在低浓度条件下可以获得SM-SERS信号, 并认为其观察到的SERS光谱波动现象可以作为确认SM-SERS的实验依据.然而在低浓度条件下, 分子落在热点区域内的概率非常低, 因此有大量的区域无法观察到任何信号, 导致SERS信号有很大的随机性.另外, 通过光谱波动确定SM-SERS事件也不严谨, 因为在多分子事件或者其他事件中也可以观察到类似现象[46,47].综合而言, 这类方法无法严格甄别单分子光谱和少分子光谱的区别, 由此得到的物理机制难免有一定瑕疵; 但这类方法为研究单分子或少分子体系SERS提供了一种简捷的途径, 特别是在动态化学反应检测中能够给予快速的信号反馈, 为分析和寻找反应中间产物提供了有效方法.
另一种获取SM-SERS的方法主要从统计学角度出发来确保获得的光谱为单分子光谱.Le Ru等[48]发现, 处于高浓度探针分子溶液中的金属纳米结构表面吸附的分子数目服从高斯分布, 而超低浓度探针分子环境中与金属结合的分子数目服从泊松分布.对泊松分布的数据进行科学分析要求SERS事件之间的强度变化小于2倍, 并且还要采集10000组以上有效数据, 这使得严谨的SM-SERS很难实现[22].为此Le Ru[29]等提出使用双分子分析测量法: 同时对两种不同的分子进行SERS检测, 并在结果中寻找只有单个分子光谱的独立事件.在高浓度样品中由于存在大量的分子, SERS信号原则上应该是两种分析物的混合光谱.然而在单分子水平中, 热点区域内只有少量分子存在,测量获取的SERS光谱会出现只有单个分析物SERS光谱的独立事件.但是从统计学角度来讲, 利用双分子分析测量法获得SM-SERS信号选用的样品浓度较高时, 获得SERS光谱为单分子事件的概率更高.这是因为采集光谱时会出现混合及纯A(或纯B)光谱事件, 混合光谱显然为多个分子(一个以上的分子)事件, 而纯A(或纯B)光谱事件可能源于两种情况: 1)检测区域内所有分子都是同一种分子; 2)或者检测区域内有单个分子位于热点区域, 掩盖了其他分子的SERS光谱, 即我们所需要的单分子光谱.在分子浓度相对较高的情况下检测区域包含的靶分子数目会增多(假设有N个),检测区域内N个分子为同一种分子(纯A或纯B分子)的概率为(1/2)N, 因此该信号为单个分子光谱的概率为1–(1/2)N.在N较高时, 此概率接近于1, 因此可以大概率确定获得的纯A(或纯B)光谱是单分子事件.这种方法从统计学角度更严谨地证实了SM-SERS的存在, 并被广泛的使用.Mao等[49]通过物理沉积的手段获得银纳米颗粒-金纳米碗复合衬底, 通过模拟计算发现该结构具有强大的电磁场增强效果.基于双分子分析方法在该基底表面成功获得SM-SERS信号.实验发现在采集了大量光谱后经过统计计算发现单个Rh6G分子事件与单个CV分子事件比例接近相同, 如图5(a)所示.该结果与先前Etchegoin等[50]报道的单分子灵敏度比率相匹配.另一种与之类似的方法使用含有某种同位素的相同分子来代替两个不同分子进行检测, 从而保证了两种分子具有完全相同的化学结合能和拉曼散射截面, 可以更准确地判断SMSERS事件[51], 如图5(b)所示.双分子分析方法虽然没有从实验角度解决“热点”不均匀分布及靶分子在“热点”中有效吸附的问题, 但是从测量方法和统计学角度给出了获取SM-SERS光谱的科学方法, 更加严谨地证实了SERS的单分子检测能力.
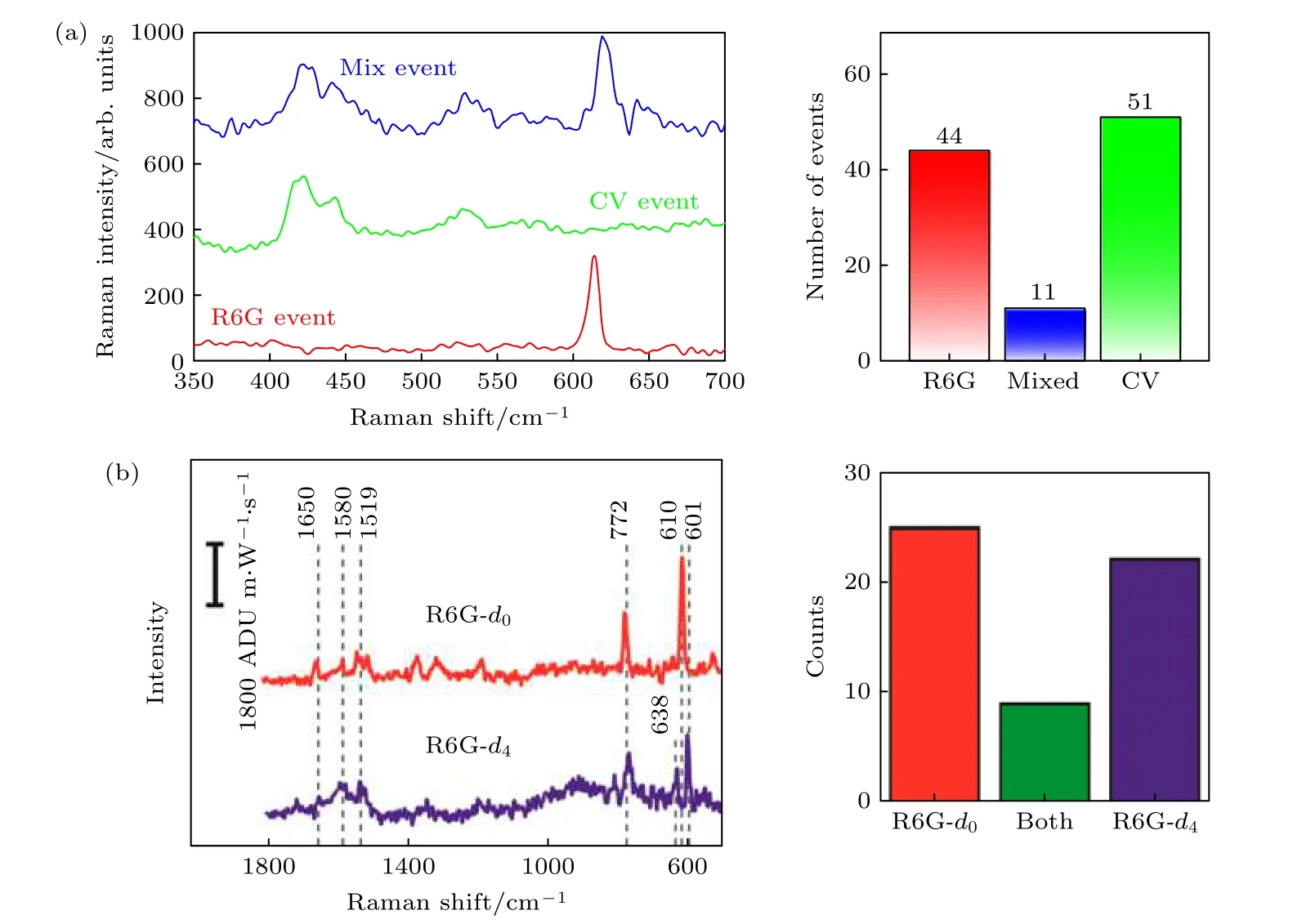
图5 双分析物测量法获取SM-SERS (a) Rh6G事件、CV事件和混合事件的SERS光谱图(左)及三种事件的统计直方图(右)[49]; (b)不同同位素标记Rh6G后获得的三种不同独立事件SERS光谱(左)及三种事件发生次数统计图(右边)[51]Fig.5.Typical SM-SERS spectra in bi-analyte measurements: (a) SERS spectra of an Rh6G event, CV event and mixed event(left), and statistical histograms of the three events (right)[49]; (b) SERS spectra of three different independent events (left), and statistics of the three events (right) by labelling Rh6G with different isotopes[51].
双分子分析方法从统计学角度给出了获取SM-SERS的方法, 然而在实验中使用了较高的分子浓度, 导致大量分子吸附在无效区域, 无法充分发挥SERS的高灵敏度的优势.因此SERS技术需要进一步对材料进行设计和优化, 降低对分子浓度的需求, 使SERS的检测限能够被真正地提高.对此相关学者提出了两类不同的解决方案, 第一类方案先将分子分散在固体基底表面, 然后在分子位置构建“热点”进行探测, 这样能够保证待测分子处于有效“热点”区域, 从而获得SM-SERS光谱.其中代表性技术为TERS[33,34], 纳米腔体增强拉曼散射[52]及厦门大学的田中群院士团队[53]提出的壳层隔绝纳米颗粒增强拉曼散射.此类技术具有纳米级甚至亚纳米级别的空间分辨率, 增强能力较高,可以探测拉曼活性较低的分子, 适用于针对特定分子进行原位研究.而第二类工作的思路与上述方法相反, 先设计材料的“热点”结构, 然后再将单个目标分子诱导至“热点”区域.常见的方法是利用表面改性、DNA生物技术等方法对热点区域进行修饰,然后将待测分子吸附或固定到“热点”位置[30,54], 该方法不需要用到双分子测量方法以及复杂的检测设备, 在实用性和经济性方面具有优势, 其技术特点有利于发挥SM-SERS特异性检测的优势, 进一步拓宽SERS在DNA组装[55]、蛋白质结构分析[56]及多相催化[57]等领域的应用.
3 单分子SERS的光谱特性与机理分析
早期研究阶段, 科研人员在寻找单分子光谱过程中将SERS信号的剧烈波动视为SM-SERS的特征.光谱波动包括: 分子的漂移, 光谱的闪烁, 以及特定振动模式的相对强度变化等.后来经过研究证实在多分子或其他环境中也会出现类似的实验现象[47].虽然这种获得SM-SERS的方法缺少科学严谨性, 但是光谱波动也确是SM-SERS的一种特征性行为.研究SM-SERS光谱波动能够从微观的角度去理解单分子的物理化学性质, SERS的增强机理及光谱本身的一些现象, 使研究者能够清楚地认识到化学反应的本质以及分子在微观系统中的行为特性.在本节中我们将从分子的漂移[58,59]、光谱强度的闪烁[60,61]及光谱展宽[21,62]等方面[63,64]讨论SM-SERS的光谱波动, 并结合相关研究报道对其中的物理机制做详细地分析.
3.1 分子漂移
在SM-SERS研究中, 分子漂移指探针分子在热点附近内外做无规则运动[65], 其主要原因可能是由于热效应, 以及分子与衬底表面之间发生电荷转移所导致[59,66,67], 而SERS中的电磁场增强是导致SERS光谱随空间和时间变化的主要原因[22,68].如6图所示的二聚体结构, 根据电磁场耦合理论二聚体纳米结构在外场Eλ的作用下, 间隙中会产生强烈的局域电磁场Eloc(“热点”), 而且随着间隙的减小(间隙不小于0.5 nm)Eloc增强[69].考虑到单个分子在“热点”附近内外做无规则运动以及相应光学理论, SM-SERS的信号ISM-SERS可以表示为[70]

其中,ωR和ω0分别表示单个分子偶极子的振荡频率以及激发光的频率,α′表示拉曼张量,G(Ω)是拉曼张量α′经变换后的旋转矩阵,Ω是探针分子的取向,rm表示分子与“热点”中心处O(电磁场最强的区域)的距离.为了更好的理解分子漂移产生的SERS强度波动, 我们用图6来解释这一原因.当单个分子吸附在热点区域内, 分子发生横向漂移(rm变化)使分子位置发生几个纳米的变化从而导致分子SERS强度发生几个量级的变化.

图6 单个分子在热点区域内发生漂移示意图, 其中Eλ是入射光的电场, K ′ 表示波矢, O是“热点”中心Fig.6.The schematic diagram of the diffusion of a singlemolecule in the hot spot area.Where Eλ is the electric field of the incident light, K ′ is the wave vector and O is the"hot spots" center.
Stranahan等[58]结合超分辨成像技术在银二聚体中绘制出SERS质心的二维分布图以及每个质心位置处的平均SERS强度.研究人员发现质心的运动是金属表面单分子发生横向漂移所导致的,并证明了单个Rh6G分子随着时间的变化在热点内外发生漂移, 如图7所示.随后该课题组利用SM-SERS同位素标记的分析物超分辨率成像揭示了SERS发射质心和相应强度之间的空间关系, 并以此说明了纳米颗粒表面上的分子运动行为[71].该实验结果有助于追踪单个分子在SERS活性纳米颗粒表面上的运动, 并且能够绘制出图像说明纳米颗粒是如何实现局域电磁场增强的.此外, 吸附在热点附近分子的取向Ω发生变化(定向漂移)也会影响光谱的波动.因为分子的吸附方式可能是平躺、倾斜或垂直于金属表面, 在这种情况下分子的偶极矩方向可能会表现出与热点中电磁场的极化方向不一致的情况, 这种差异在SM-SERS光谱中可以明显被观察到.Marshall等[72]基于实验测量和模拟计算证明该研究中所制备的银纳米球腔体具有SM-SERS检测能力, 并发现来自纳米间隙的SM-SERS信号强烈依赖于分子取向.在单分子体系中, 分析物的随机取向不再是许多分子的平均,因此必须考虑分子拉曼活性偶极矩相对于增强电场极化的方向.结合密度函数理论计算结果与光谱实验, 作者确定了等离激元纳米间隙中分子的取向分布情况, 如图8所示.这种在单分子水平上对分子取向的研究和确认对于分子电子器件方面的研究具有重要借鉴意义.该研究团队随后继续在这一纳米系统中研究了单分子级别共轭聚合物分子的取向分布情况[73].结合理论计算和SM-SERS光谱数据得出共轭聚合物分子链相对于金属表面更容易呈现平行吸附模式.该实验为理解共轭聚合物在金属表面的取向分布, 优化共轭聚合物器件(如: 有机发光二极管等电子器件)提供了可靠的实验支撑.

图7 单个分子漂移行为的探究[58] (a) SERS质心位置在二维空间中出现的次数(右边色条代表事件次数); (b) 在每个质心位置测得的平均SERS强度, 其中纳米粒子发射质心设置为(0, 0); (c) SRER质心的位置和强度随时间发生变化, 这表明单个探针分子在热点附近发生漂移Fig.7.Study on the diffusion behavior of a single molecule[58]: (a) The number the SERS centroid position that appears in two-dimensional space during the detection (the right color bar represents the number of events); (b) the average SERS intensity measured at each centroid, where the emission centroid of nanoparticles is set to (0, 0); (c) the intensity and position of SERS centroid change with time, which indicates that single molecule wanders around the hot spots.

图8 SM-SERS中分子不同取向对光谱产生波动影响[72] (a) 银纳米腔体结构示意图; (b) 光谱1—3表示纳米间隙中不同取向分子的SM-SERS信号, 光谱4表示高浓度普通SERS光谱; (c) Rh6G分子的结构以及不同场方向在分子α-γ和α-β平面上的投影Fig.8.The effects of molecular orientations on spectrum fluctuations in SM-SERS[72]: (a) Schematic diagram of silver nanocavity structure; (b) spectrum 1–3 is SM-SERS signals of molecules with different orientations in the nan-gap, and spectrum 4 is the SERS spectrum of high concentration molecules; (c) the chemical structure of Rh6G molecule and the projection of different field directions on the α-γ and α-β planes of this molecule.
SM-SERS的另一个重要应用在于对催化反应内在机理的研究, 催化反应涉及复杂的反应步骤和多样的中间产物, 实验中往往只能观察到最终产物或几种重要的中间产物, 而对大多数的中间过程无法进行系统地研究.SM-SERS技术可以对化学反应过程进行实时监测, 能够对反应过程的中间产物结构及反应步骤进行化学反应动力学分析, 有助于研究人员从微观状态研究和认识化学反应的本质.但是在实际操作中, 对于给定的SM-SERS轨迹中的单个事件, 研究人员无法确定该事件是由化学反应引起的还是因为单个分子发生漂移或者其他非反应过程所导致.基于此, Choi等[65]通过在“银纳米球-金膜”结构的纳米间隙中吸附4-NTP分子对这一问题进行了研究, 如图9所示.在波长为633 nm的激发条件下, 作者观察到了随时间变化并且具有阶跃特征的SERS光谱, 并认为这些光谱的阶跃信号对应于单个分子在热点处发生的化学反应事件.由于单个分子在热点中发生漂移时只涉及物理变化而与化学反应无关, 因此光谱的阶跃频率f与分子的衰减速率常数kNBT之间不存在线性关系.然而, 作者在该实验中发现4-NTP分子的衰减速率常数kNBT与光谱的阶跃频率f呈明显的线性依赖关系, 如图9(d)所示.因此, 作者认为实验中观察到的光谱波动现象是由化学反应所导致, 从而排除了分子发生漂移的可能性.

图9 (a)“银纳米球-金膜”纳米腔体结构用于SERS测量及纳米腔暗场散射光谱[65]; (b) 催化反应生成中间产物的间歇性光谱特征轨迹[65]; (c) 在开(红)和关(蓝)期间从图9(b)中取样的SERS光谱, 两者光谱的差异(橙)及其协方差矩阵VX = 1364 cm–1分量(绿).在1160, 1314, 1364 cm–1处出现间歇性SERS峰归因于中间产物羟胺基苯硫醇分子的拉曼峰[65]; (d)光谱的上升沿阶跃频率fup(左)和下降沿阶跃频率fdown(右)与4-NTP分子的衰减速率常数kNBT之间的关系[65]Fig.9.(a) The “silver nanosphere-gold film” nanocavity structure in SERS measurement[65] (This figure contains chemical structures of NBT and DMAB molecules, as well as the dark field scattering spectrum of a single nanostructure cavity); (b) the trace of the intermittent spectral characteristics of intermediates generated by catalytic reaction[65]; (c) SERS spectra sampled from Fig.9(b)during on (red) and off (blue), the differences between the two spectra (orange) and the VX = 1364 cm–1 component (green) of the covariance matrix.The intermittent SERS peaks at 1160, 1314 and 1364 cm–1 are attributed to the Raman peaks of the intermediate product hydroxyl aminobenzenethiol[65]; (d) the relationship between the spectral step-up frequency fup (left) and step-down frequency fdown (right) and the decay rate constant kNBT of 4-NTP molecule[65].
基于上面的讨论分析, 我们总结出在SM-SERS中分子的漂移分为两种情况: 横向与定向.对于横向漂移而言, 由于分子在热点内外做无规则随机运动, 分子经历了不同强度的局域电磁场使得SERS强度发生变化.在定向的漂移中, 分子在金属表面的吸附方式不同造成分子的取向与局域电磁场极化方向存在差异, 导致拉曼峰的相对强度发生变化.在SM-SERS信号采集实验中, 这两种漂移方式有可能是同时存在的, 并且随着光谱采集时间的推移, 我们也会从光谱数据中发现分子在某些波数处的拉曼峰强会出现类似“强-弱”交替的循环现象, 这就是接下来要讨论的SM-SERS中的“闪烁”现象.
3.2 光谱闪烁
一般在采集SM-SERS光谱时会发现探针的拉曼信号在某个波数处发生波动, 该现象被称为闪烁, 它是SM-SERS中光谱信号发生“开-关”循环并且可以持续几毫秒甚至几分钟的一种实验现象.通常认为分子发生漂移是SM-SERS发生闪烁的根本来源.当金属纳米结构的形貌、颗粒之间的间距及激发波长等外在实验条件确定后, 衬底的热点强度和分布不会发生明显地变化, 但是处在热点中的单个分子会在热点内外产生漂移, 导致该单个分子经历不同强度的电磁场(几个数量级的变化)[22],这是SM-SERS中闪烁现象的根本原因.随着SMSERS研究进一步的深入, 有研究表明温度升高产生的热效应[74−77]、电荷转移[61,67]及其他因素[61,78]也是诱导单分子闪烁的原因.下面将对SM-SERS闪烁背后的基本理论和相关工作做具体讨论.
单分子光谱波动的实验现象一直存在.在光谱测量过程中, 不管是拉曼信号还是荧光光谱信号都会出现这样的情况.为了更清晰地揭示该现象背后的物理机制, Haran工作组[78]在银纳米晶体上吸附Rh6G分子, 并在532 nm光源的激发下观察到了单个Rh6G分子在数秒级的时间尺度上发生了闪烁现象, 如图10所示.作者研究了不同条件下单个Rh6G分子的SERS光谱动力学, 并进行了统计分析.研究发现, 单个Rh6G分子的SERS信号随时间波动的尺度取决于激光的功率(非光热)和电解质的浓度.随着激光功率的提高, 光谱波动尺度增大.作者认为其主要原因是由金属与吸附分子之间的电荷转移所导致的.此外, 另一种解释是由于分子在金属表面运动所致.而这种运动可能是源于单个分子与金属表面发生了“脱吸-再吸附”的现象.然而, 随着胶体中盐溶液浓度的增加, 胶体颗粒周围双层的静电势发生变化, 影响金属表面的局域功函数(局域电荷密度), 从而导致SM-SERS的光谱波动被强烈抑制.该工作为后续研究SMSERS中的闪烁现象提供了理论依据.

图10 SM-SERS的闪烁现象[78]: 单个Rh6G分子SERS信号随时间变化的轨迹图, 其中在波数614 cm–1和774 cm–1处发生明显的光谱波动Fig.10.The Blinking phenomenon in SM-SERS [78]: The time-dependent trajectories of SERS signal of a single Rh6G molecule.Obvious spectral fluctuations are observed at 614 cm–1 and 774 cm–1.
Maruyama等[76]通过实验证明了温度升高产生的热活化效应会导致SERS光谱产生闪烁现象.实验发现SM-SERS光谱闪烁现象与温度有关.当温度变化时, 研究者观察到了拉曼光谱闪烁现象的发生-抑制-复现.考虑到热效应可能对金属纳米颗粒形貌有影响, 作者通过改变激光功率及在高温环境中退火1 h后, 发现金属的形貌没有发生明显的变化, 排除了局域电场变化对探针分子信号的影响.因此, 作者认为温度升高产生的热效应导致单分子拉曼信号发生闪烁现象.这可能是由于Ag表面等离激元热效应提供了足够大的热能克服了分子的扩散势垒, 使得分子能够在金属表面发生漂移.随后聂书明课题组[61]也研究了SM-SERS中闪烁的现象, 提出闪烁是由单个分子在纳米颗粒表面上的热活化扩散引起的, 并认为激光的功率、波长、光诱导的电子转移及表面活性位的结构弛豫可能是导致闪烁的原因.最近, Lindquist[79]等通过快速成像技术发现金属纳米结构表面的原子扩散引起热点变化也是导致SM-SERS发生闪烁现象的一个原因.研究人员利用一种捕获率为800000帧/s并且空间分辨率为7 nm的成像技术对银纳米壳层表面单个分子的拉曼光谱进行探测, 如图11所示.实验发现在金属表面被化学键固定的分子的SERS光谱信号以一定的速度在发生波动.作者通过进一步的观察和研究推断出金属表面银原子的重构驱动热点产生移动是导致SERS波动的原因, 其模拟分析结果与推断结论几乎一致.
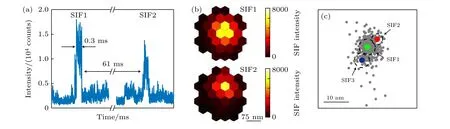
图11 高速SERS信号采集和超分辨率成像[79] (a) SM-SERS放大后的两个波动图, 每个持续约0.3 ms, 相距61 ms; (b)光电探测器上接收到两个易于区分的光谱波动事件成像; (c)在纳米粒子的空间位置上绘制的三个光谱波动轨迹Fig.11.High-speed SERS acquisition and super-resolution SERS imaging[79]: (a) Each of two amplified wave patterns of SM-SER spectra lasted for about 0.3 ms and separated by 61 ms; (b) images of spectral fluctuation events; (a) three spectral wave trajectories on the spatial position of nanoparticles.
通过上述典型的研究报道可以将SM-SERS发生闪烁的原因归结如下: 1)由于热效应导致热点附近的单个分子发生漂移扩散; 2)分子与金属表面存在电荷转移; 3)在溶液体系中由于分子做无规则运动导致分子在纳米结构表面发生脱吸和再吸附的过程; 4)染料分子发生光漂白; 5)热点扩散.虽然与闪烁相关的研究工作较多, 但是该研究仍是一个有争议的话题.目前大多数研究只是通过实验定性地分析了闪烁的原因, 因此仍然需要做进一步工作从实验现象背后的物理机制来分析该现象.在采集SM-SERS光谱时, 分子的闪烁是我们要注意的一个现象.因为通过SM-SERS光谱研究单分子化学反应过程需要区分到底是反应导致的光谱发生波动还是发生了闪烁现象, 这就要求我们选择合适的激光功率和波长、温度及适宜的环境体系等来避免闪烁现象对实验的影响.
3.3 光谱频移与展宽
在拉曼光谱中可以观察到均匀展宽和非均匀展宽的现象, 前者源于分子内的非谐相互作用, 这是单分子振动的一个基本特性, 而后者则代表了分子在样品中不同位置中的变化[21].在收集SM-SERS光谱时, 研究者发现这些单个分子的光谱拟合成平均光谱后, 光谱呈现出不对称的洛伦兹曲线, 这种现象叫做SM-SERS光谱的不均匀展宽现象.另外, 随着温度的升高SM-SERS光谱的谱宽表现出增大趋势, 该现象称之为SM-SERS光谱的均匀展宽.在SM-SERS中, 分子间的相互作用及分子与周围环境的相互作用都会导致光谱出现展宽现象.
Etchegoion等[62]在低温环境下, 利用高分辨光栅(2400 mm–1)观察了尼罗蓝分子和罗丹明800分子分别在590 cm–1和2226 cm–1处的不均匀展宽现象, 如图12(a)和12(b)所示.作者拟合了大量的单分子光谱后发现, 与正常的多分子光谱相比, 拟合的平均光谱呈现出不对称分布的现象.作者认为不均匀的展宽是由于分子间的相互作用或分子在底物上的相互作用引起的, 并且单个分子光谱之间拉曼峰的频率差异是导致非均匀展宽的主要原因.此外, 作者从分子与基底的相互作用,同位素效应, 还有仪器方面解释和讨论了单分子光谱频率发生移动的可能来源.研究结果表明同位素位效应、光谱仪响应不完全是频率漂移的主要原因.对同一分子不同峰的不均匀展宽进行数据统计分析将有助于揭示和理解分子与周围环境相互作用的具体行为细节.随后该课题组[21]又研究了温度变化对单个尼罗蓝分子拉曼增强光谱在590 cm–1处的均匀展宽现象的影响.随着温度的升高单分子光谱的半高宽发生均匀展宽现象(半高宽大约从0.9 cm–1增加到2.7 cm–1附近), 如图12(c).作者认为随着温度升高光谱出现均匀加宽的现象是由于分子与基底银声子发生非简谐耦合所导致.研究SM-SERS的光谱展宽现象, 有助于进一步了解分子与其周围环境的相互作用(分子之间或分子与基底之间)及温度等外在因素对分子自身行为的影响.
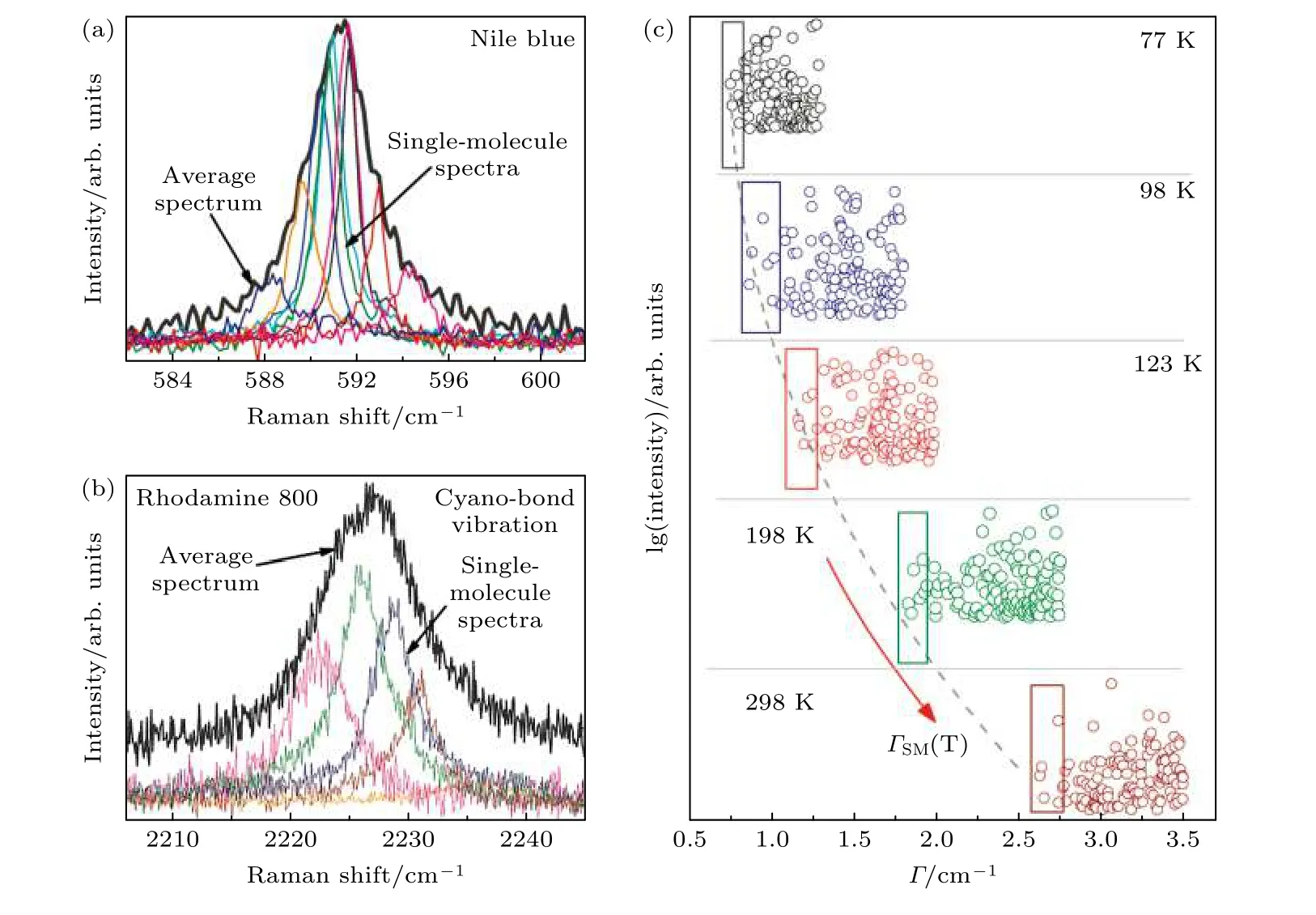
图12 SM-SERS光谱的展宽现象 (a)单个尼罗蓝染料分子在590 cm–1能量振动处的不均匀展宽现象[62]; (b)单个罗丹明800分子在2226 cm–1能量振动处的不均匀展宽现象[62]; (c) SM-SERS事件中单个尼罗蓝分子在590 cm–1处光谱的半高宽(Γ)与温度(T)之间的关系, 表明随着温度T升高SM-SERS光谱发生均匀展宽[21]Fig.12.Broadening of SM-SERS spectrum: (a) Inhomogeneous broadening of a single Nile Blue dye molecule at 590 cm–1 energy vibration[62]; (b) inhomogeneous broadening of a single Rhodamine 800 molecule at 2226 cm–1 energy vibration[62]; (c) the relationship between the FWHM (Γ) of a single Nile Blue molecule indifferent SM-SERS events at 590 cm–1 with increasing temperature T. The result indicates that the SM-SERS spectrum broadens uniformly with the increase of temperature (T)[21].
3.4 小 结
拉曼光谱信号波动现象是SM-SERS光谱最明显且重要的特征.虽然研究人员当前已经从温度、激发条件、电荷转移等影响因素分析了SMSERS光谱波动的原因, 但是如果要深究其背后的理论机制及相关的应用仍然需要做进一步的工作.目前对该系统的研究和理解尚不够全面, 而单分子纳米级别的系统中是一个复杂的状态, 并且影响光谱波动的因素还可能与分子本身及分子所处的环境有关, 如: SM-SERS的振动涌出现象及高压环境诱导SM-SERS峰位发生蓝移现象等[64,80,81].因此, 在SM-SERS光谱波动的研究中需要尽可能设计合理巧妙的实验方法来排除其他因素的干扰.目前已经有报道通过在金属表面构造超薄的壳层来避免电荷转移对光谱波动的影响[53]; 而且研究人员利用物理、化学及生物等手段制备出的超灵敏检测基底已满足SM-SERS的检测灵敏度, 但是如何对单个分子进行精确地操控使靶分子能够有效吸附到“热点”区域并减弱或者排除单个分子光谱波动对分子动力学研究的干扰影响, 仍然有待进一步的讨论和分析.
4 总结与展望
本文重点研究和探讨了SM-SERS的信号的获取方法, 以及SM-SERS的光谱波动特性和相关机理.SM-SERS的优势在于其对分子及亚分子化学键的特异性识别能力, 这项技术已经被初步应用于监测催化反应过程[44,45,57,82]及生物分子测序[55,56,83,84]等领域.此外, SM-SERS对化学键特异性识别的能力也使得研究分子与周围环境的电荷相互作用成为可能, 这被应用于确定分子的吸附构型、吸附取向分布[36]; 研究分子在光激发作用下的解吸附过程[67]; 观察分子与基底之间的电荷转移等[85].另一方面, 随着表征仪器不断的发展, 近年来研究者人员在SM-SERS检测中结合TERS技术能够在单分子水平中区分结构相似的待检测分子, 其亚纳米级别的空间分辨率可以显示出单个分子的分布状态、振动模式及吸附方式等重要信息, 该技术有望被推广到单分子成像研究领域[33−38,86−88].
然而, SM-SERS在信号采集和数据处理方面需要更加可靠的技术和严谨的方法, 除了本文已提到的部分, 还需要注意: 1)在SM-SERS光谱收集实验中, 激发条件、温度、光栅分辨率及光谱仪中CCD的曝光时间等因素会导致光谱波动或影响光谱采集效果.所以在研究分子动力学的过程中, 需要考虑并优化实验方法排除这些因素对实验结果的影响.2)双分子分析方法假设每个纳米间隙之间形成的“热点”都会提供等效的增强因子.而实际上纳米颗粒之间的间隙以及形貌难以完全统一, 这种假设在实验中很难实现.因此, 需要建立更加完善和健全的统计方法使SM-SERS检测更具有科学合理性.3)分子总是处于变化且复杂的状态, 揭示单个分子的自身行为, 以及与周围环境的相互作用时要考虑到瑞利散射和量子效应等其他的物理效应[19].4)在TERS技术中, 如何能够稳定、有效地制备出具有几个金属原子组成的超高局域电磁场分布并实现单分子检测的针尖仍然是一个挑战.
本文从SM-SERS的内涵, SM-SERS信号的获取及SM-SERS的光谱特征等方面进行了初步的探讨.由于SM-SERS技术能够提供极高的灵敏度及单个分子在待测环境中的行为特征, 因此该技术有望用于单分子表征分析等相关领域.希望本文能够引起读者的兴趣和对本领域的关注, 在机理分析和实验构筑等层面上进一步拓展和丰富SMSERS的相关研究.
