一种燃料热沉标准物质候选物的纯度定值及不确定度分析
2021-08-03王志轩杜咏梅李春迎孙道安李娇毅
王志轩,徐 强,杜咏梅,李春迎,孙道安,李娇毅,张 皋,吕 剑
(西安近代化学研究所 氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065)
1 引 言
高超声速(≥5倍声速)飞行器具有突防快、难拦截的优势,代表着国家武器装备的发展水平,是当今世界各大国优先发展的高新技术之一[1~3]。燃料的热沉(即特定工况下的吸热能力)直接决定了飞行器的速度,如燃料的热沉达到2.09 MJ/kg时,可满足飞行器4~6马赫的飞行速度需求[4,5]。准确、高效地计量燃料热沉对高超声速飞行器的设计及吸热燃料研制至关重要。然而目前我国在热沉标准物质研制方面处于起步阶段,因而难以为吸热型碳氢燃料在高超声速飞行器中的工程应用提供热沉计量保障。为保证热沉测量结果的一致性、可比性及可溯源性,急需开展相应的热沉标准物质的研制工作。
一种具有多环结构的烃类化合物,是目前吸热型碳氢燃料JP-7和RP-3等的关键组分。并且,它具有来源广、毒性低或无毒、热稳定性高、吸热能力强等特性,作为热沉标准物质候选物(以下简称候选物)极为适合。从吸热型碳氢燃料化学组成角度出发,纯度对其特性量值热沉影响显著。
目前,针对有机化合物标准物质的纯度定值方法,主要有质量平衡法、核磁定量法和示差扫描量热法等[6~11]。其中,质量平衡法是一种间接的纯度测定方法,它不仅可以准确定量有机标准物质的纯度,而且可以同时对有机杂质、水分、无机杂质等含量进行定量[12,13]。核磁定量法测定快速准确、专属性强、不破坏样品,样品预处理较为简单。通过选择有证标准物质作为内标物,可以将测定结果直接溯源到有证标准物质上,从而为测量结果提供了溯源保障,该方法是目前有机物质纯度定值的常用方法[14,15]。
综上,本文采用质量平衡法和核磁定量法2种不同原理的纯度定值方法,对研制的标准物质候选物的纯度进行定值,并对其不确定度进行了分析和评定。本研究可为后续燃料热沉标准物质的研制和申报奠定一定的基础。
2 实验部分
2.1 仪器与试剂
气相色谱-质谱联用仪GC/MS-QP2010Plus(GC-MS),日本岛津公司;卡尔-费休库仑法水分测定仪C20S,瑞士梅特勒公司;燃料胶质含量测定仪FDR-0531,长沙富兰德公司;电感耦合等离子体质谱仪ICP-MS7700,安捷伦公司;喷气燃料固体颗粒污染物测定仪KFD-R2801,北京中西科仪器科技有限公司;电子分析天平BSA224S,赛多利斯公司;核磁共振仪BrukerAV500,德国Bruker公司。
卡尔-费休试剂,北京百灵威科技有限公司;候选物,实验室自制;乙酰苯胺标准物质(编号:71168, 批号:0381907, 纯度:99.8%, 不确定度:0.5%)坛墨质检科技股份有限公司;氘代氯仿(99.8%),北京百灵威科技有限公司。
2.2 实验方法
2.2.1 主组分测定
气相色谱测定主组分及挥发性杂质方法:DB-35色谱柱(30 m×0.32 mm×0.50 μm),载气流速为 1 mL/min,进样量为1 μL,分流比为20:1,顶空进样,进样口温度为280 ℃,起始柱温为50 ℃,保持15 min,以15 ℃/min程序升温至280 ℃,保持 5 min, FID检测器温度为280 ℃。
2.2.2 水分测定
采用卡尔-费休库仑法水分测定仪,依国标GB/T606《化学试剂水分测定通用法-卡尔费休法》测定。
2.2.3 非挥发性有机杂质测定
采用喷射蒸发法胶质含量测定仪,依据GB/T 8019-2008《燃料胶质含量的测定喷射蒸发法》测定。
2.2.4 无机杂质测定
采用电感耦合等离子体发射光谱仪,依GB/T 17476-1998(2004)《使用过的润滑油中添加剂元素、磨损金属和污染物以及基础油中某些元素测定法(电感耦合等离子体发射光谱法)》测定。
2.2.5 固体颗粒污染物测定
采用液体燃料固体污染物测定仪,依据石油化工行业标准SH/T 0093-1991《喷气燃料固体颗粒污染物测定法》测定。
2.2.6 核磁定量法纯度测定
核磁共振仪:1H NMR,500 MHz(德国Bruker公司),测定参数:激发脉冲角度为30 ℃,采样时间为3.276 9 s,扫描宽度为10 000.000 Hz,弛豫延迟1 s,累计采样64次,探头温度299.4 K,偏置频率为0.152 588 Hz,接收增益为90.5,脉冲序列为zg30。
采用标准物质乙酰苯胺(99.8%)为内标物,将15 mg的标准物质候选物与6 mg乙酰苯胺内标物共同溶解于1.0 mL氘代氯仿溶剂中,待充分溶解后,移取0.5 mL至石英核磁管中待测,同时将标准物质候选物与乙酰苯胺内标物分别溶于氘代氯仿中作为参照样品来确定化学位移的归属。
3 结果与讨论
3.1 定性分析
本文选用实验室自制的一种多环烃化合物作为燃料热沉标准物质候选物,对候选物的结构定性的方法和过程详述如下。
在候选物结构定性过程中,首先采用GC-MS对标准物质候选物进行了结构分析。通过候选物的分子离子峰和特征碎片离子峰,结合化合物的分子结构特点,初步确定实验室研制的候选物即为所需的标准物质候选物分子结构。
为进一步确认其分子结构,采用核磁共振氢谱和碳谱分别对候选物进行了结构鉴定。依据氢谱中氢原子个数及其化学位移、碳谱中碳原子个数及其化学位移相关信息,并结合GC-MS的分析结果,可确认标准物质候选物候选物的分子结构。在此基础上,本文筛选出适合定量分析的乙酰苯胺作为内标物,对候选物的纯度进行了核磁定量分析,相关结果见第3.2.4节。
3.2 定值分析
采用质量平衡法与核磁定量法2种不同原理的方法对候选物进行纯度定值分析。
3.2.1 质量平衡法
候选物是由石油副产品萘经过加氢、异构化、精馏分离、精制纯化而来,其杂质主要可分为水分、无机杂质(主要考虑金属离子类)、挥发性有机杂质、非挥发性有机杂质(即胶质)和固体污染物(主要包括灰分、机械杂质等悬浮物)等。
由于候选物沸点约185 ℃,为挥发性有机物,因此本文中气相色谱法(GC-FID)可同时测定候选物主组分和挥发性有机杂质色谱纯度,且其和为100%。水分采用卡尔费休库伦法测定,非挥发性有机杂质胶质采用GB/T 8019-2008测定,无机杂质采用GB/T 17476-1998(2004)(ICP-OES法)测定,非溶性固体污染物采用SH/T 0093-1991测定。
质量平衡法是通过分别测定纯品有机物中水分(Xw)、挥发性有机杂质(Xo)、非挥发性有机杂质(Xno)、无机杂质(Xi)和非溶性固体污染物(Xs)等,计算目标成分含量(P)的方法。如前所述,气相色谱法中主成分和有机挥发性杂质含量和为100%,因此质量平衡法中纯度标准物质含量计算方法如下:
P=1-Xw-Xo-Xno-Xi-Xs
(1)
3.2.2 质量平衡法不确定度分析
根据质量平衡法的内涵,其纯度定值不确定度分量主要有水分、挥发性有机杂质、非挥发性有机杂质、固体污染物等测量引入的不确定度。其中各分量重复性测量数据列于表1,不确定度分析和评定具体过程如下:

表1 质量平衡法测定结果
(1) 挥发性有机杂质测量引入的不确定度
挥发性有机杂质采用气相色谱法定量,其引入的不确定度u(Xo)主要有3个分量:1) 测量重复性产生的不确定度u1(Xo)。本实验中平行测量6次,计算得到相对不确定度u1rel(Xo)=0.003 1%。2) 组分校正因子差异引起的不确定度u2(Xo)。由于本纯度标准物质候选物挥发性有机杂质主要是候选物的同分异构体和结构相近的双环化合物,因此,其响应因子与主成分理论分析十分接近。依据气相色谱分析结果,挥发性有机杂质总含量为0.51%。预估总响应因子差异引入的不确定度为杂质总含量的30%,则相对不确定度u2rel(Xo)=0.15%。3) 检测线性引入的不确定度u3(Xo)。由于纯度定值方法研究过程中所确定的进样量均在FID检测器的检测线性范围内,故仪器检出限引起的不确定度u3(Xo)可忽略不计。
由于上述的不确定度分量相互独立,互不相关,因此合成得到相对不确定度
=0.15%。
(2) 水分测量引入的不确定度

(3) 非挥发性有机杂质测量引入的不确定度
非挥发性有机杂质测量引入的不确定度分量u(Xno)主要有3个:1) 测量重复性产生的相对不确定度u1(Xno)。实验中平行测量6次,计算得到u1rel(Xno)=0.000 02%,可忽略不计。2) 温度产生的不确定度u2(Xno)。实验中温度分量扩展不确定度为0.20 ℃(k=2),实验温度为230 ℃,求得u2rel(Xno)=0.043 5%。3) 电子天平称量产生的不确定度u3(Xno)。实验中称量用天平相同且称量的样品质量为53.000 0 g,因此u3rel(Xno)=(0.290/53 000.0)×100%=0.000 5%。以上3个分量相互独立,互不相关,最终求得urel(Xno)=0.043 5%。
(4) 固体污染物测量引入的不确定度
固体污染物测量引入的不确定度u(XS)主要分量有3个:1) 测量重复性引入的不确定度u1(XS)。实验中平行测量6次,计算得到u1rel(XS)<0.000 1%,可忽略不计。2) 电子天平称量引入的不确定度u2(XS)。实验中称量用天平相同且称量粘附固体污染物的滤纸样品质量平均值为164.9 mg,u2rel(XS)=(0.290/164.9)×100%=0.175 9%。 3) 体积称量引入的不确定度u3(XS)。实验量取样品的体积为5 L,最大允差为±0.01 L,求得u3rel(XS)=0.2%。以上3个分量相互独立,最终求得urel(XS)=0.266 3%。
(5) 无机杂质测量引入的不确定度
采用电感耦合等离子体发射光谱仪测定的无机杂质含量极少。与其他杂质含量相比,对纯度标准物质的定值影响可忽略不计。
综上,由于质量平衡法引入的各不确定度分量相互独立,互不关联,因而将各不确定度分量进行合成,得到质量平衡法引入的相对标准不确定度:
uMB,rel=[urel(Xo))2+(urel(XW))2+
3.2.3 核磁定量法
核磁定量法原则要求内标物为有证标准物质,以实现测量的溯源性。但乙酰苯胺标准物质目前并不是有证标准物质。作者选用乙酰苯胺作为核磁定量内标物主要基于以下考虑:
(1) 该标准物质生产厂家已经取得了标准物质生产者CNAS认证,且已建立乙酰苯胺标准物质规范(企标)。
(2) 作为内标物定量候选物,其中一条关键的原则是选用的内标物在定量过程中对候选物上相关C/H元素无干扰,分离完全。作者目前虽已尝试过几种有证纯度标准物质,但均存在与待测物候选物相互干扰的现象,因此无法采用。
为此,作者暂将标准物质乙酰苯胺作为内标物,进行候选物的核磁定量。核磁定量法的定值计算公式为:

(2)
式中:PNMR为采用核磁共振法测得的样品纯度;Ix为样品指定峰的积分面积;Istd为内标物指定峰的积分面积;nstd为内标物指定峰的核群核个数;nx为样品指定峰的核群核个数;Mx为样品的相对分子质量;Mstd为内标物的相对分子质量;mstd为内标物的样量;mx为样品的秤样量;Pstd为内标物的纯度。
3.2.4 核磁定量法不确定度分析
核磁定量法不确定度的来源主要包括核磁共振仪测量不确定度、天平称量不确定度、内标物纯度不确定度以及分子量不确定度[16,17]。各不确定度分量评定如下:
(1) 核磁共振仪测量不确定度
核磁共振仪测量结果的标准不确定度u(Ix/Istd)主要由核磁仪器积分测定的不确定度造成,可用核磁定量法测定结果的平均值实验标准偏差表示。表2列出了核磁定量法平行测量结果,由表2可知,核磁共振仪测量引入的相对不确定度urel(Ix/Istd)=0.032 12%。
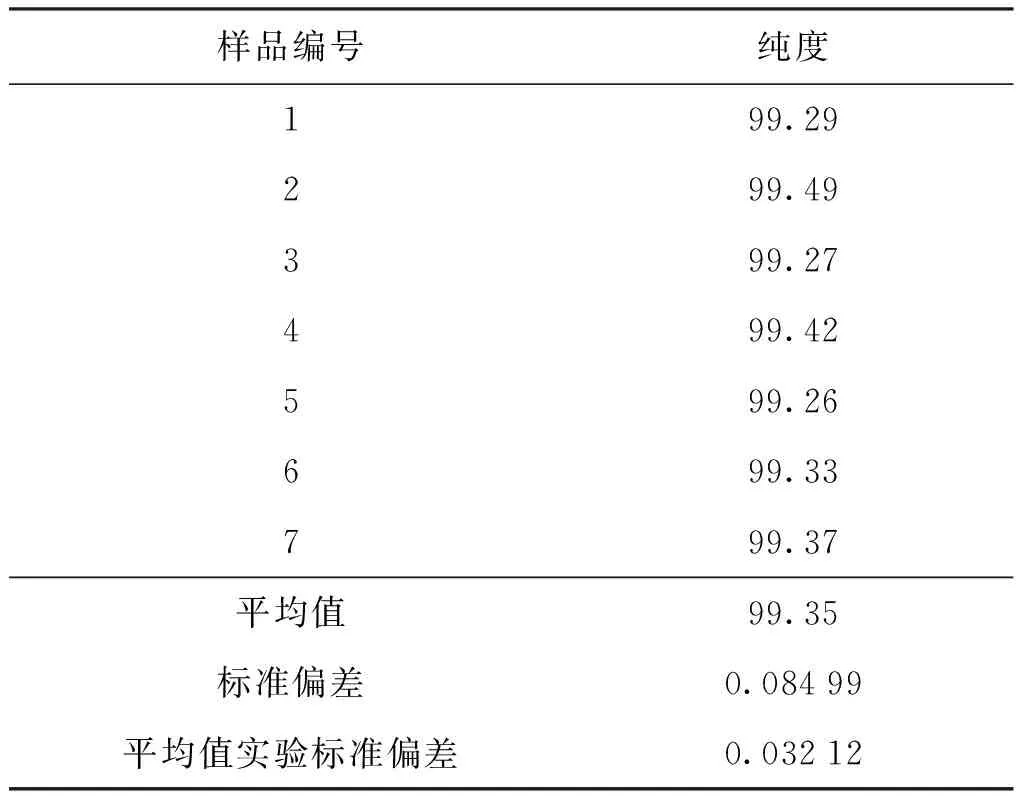
表2 核磁定量法测定结果Tab.2 Measured results of quantitative nuclear magnetic resonance method (%)
(2) 天平称量不确定度
本文中实验称样量为21 mg,求得天平称量产生的相对不确定度urel(m)=0.14%。
(3) 内标物纯度不确定度
内标物乙酰苯胺的纯度不确定度u(Pstd)由标准物质证书获得。由证书可知乙酰苯胺的扩展不确定度为0.50%(k=2),则取其相对标准不确定度urel(Pstd)=0.25%。
(4) 分子量不确定度
根据IUPAC国际原子量表,4种原子的相对原子量及不确定度为:H:1.007 94±0.000 07, C:12.010 7±0.000 8, N:14.006 74±0.000 07, O:15.999 4±0.000 3。 对于每个元素,其标准不确定度是由均匀分布的引用不确定度转化而来。候选物的分子式为C10H18,相对分子量为138.25,标准不确定度为:

=0.001 47
候选物相对分子量的相对不确定度为:urel(Mx)=u(Mx)/Mx=0.001 064%。内标物乙酰苯胺(C8H9NO)相对分子量为135.16,其相对不确定度为:urel(Mstd)=u(Mstd)/Mstd=0.000 979 6%。
核磁定量法测定纯度的相对不确定度为:

3.3 均匀性检验及不确定度分析
候选物常温下为高纯度均相液体化合物,因此在均匀性检验时,重点对分装过程中瓶间的均匀性进行了检验。具体过程为在参考相关技术规范[7]的基础上,随机从封装的标准物质候选物中抽取10个独立包装,从1到10进行编号,采用气相色谱法,以样品纯度值为特性量参数,每个单元重复测定3次,进行瓶内和瓶间均匀性检查。测量的数据采用单因素方差分析法(F检验)进行统计检验,具体结果见表3。由表可知,计算的F值为1.07,查F检验临界值表得F0.05(9,20)=2.39,即F 表3 候选物均匀性检验数据Tab.3 Homogeneity test data of candidate (%) 均匀性引入的相对不确定度ubb,rel可由以下公式计算: =0.002 6% 为考察候选物在长期储存条件下,其物理化学性质和特性量值保持不变的能力,本研究在参考《标准物质定值原则与统计学原理》的基础上,结合今后实际应用情况,采用GC-FID法对候选物纯度开展了长期稳定性考察,即按先密后疏的原则,分别在第1、2、4、8、12月时检测原料的长期稳定性。每次取3个样品,每样分析3次,以测量平均值作为检测结果。分析结果用趋势分析法(t检验法)评价,即根据时间和纯度值拟合线性回归曲线,用该曲线斜率的显著性评估其稳定性。图1给出了标准物质候选物长期稳定性考察结果。经拟合得到拟合方程为 图1 候选物稳定性检验数据Fig.1 Stability test data of candidate Y=99.474 9-0.001 27X, |b1|=0.001 27, s(b1)=0.001 73。 经查t-检验临界值表得 t(0.95,n-2)=2.447, |b1| 斜率估计值不显著,表明当置信水平为0.95时,标准物质候选物的纯度特性量值具有优异的储存稳定性。根据稳定性引入的相对不确定度的评定计算公式,求得(取12月时的数值): usts,rel=(s(b1)×X/100)/Y=0.020 87% 根据质量平衡法和核磁定量法测出的标准物质候选物纯度平均值分别为99.49%和99.35%,由于相差较小,本文取其算术平均值作为纯度值,即99.42%。 标准物质候选物的标准不确定度uCRM由3部分组成,即定值方法引入的标准不确定度uchar,包括重量平衡法和核磁定量法引入的标准不确定度;标准物质均匀性引入的标准不确定度ubb;稳定性引入的标准不确定度usts。因三者相互独立,互不关联,则合成相对标准不确定度计算如下: 相对扩展不确定度Urel=kuCRM,rel=2×0.42%=0.84%,k=2。 综上,标准物质候选物的纯度和扩展不确定度分别为99.42%和0.84%(k=2)。 (1) 核磁定量法中乙酰苯胺标准物质目前尚不是有证标准物质;但通过本研究,表明该标准物质适用于核磁定量法中对候选物纯度定值时作为内标物。因此为实现核磁定量法候选物纯度定值的溯源性,下一步将通过系统研究,使乙酰苯胺标准物质成功申报为有证标准物质。 (2) 采用质量平衡法和核磁定量法对标准物质候选物进行了纯度定值,两种定值方法定值结果基本一致。 (3) 研制的标准物质候选物具有良好的均匀性和稳定性,在常规条件下储存时间至少1年以上。 (4) 标准物质候选物定值结果的标准值和相对扩展不确定度分别为99.42%和0.84%(k=2)。
3.4 稳定性检验及不确定度分析

4 标准物质候选物纯度及不确定度分析

5 结 论
