Anomalous bond-length behaviors of solid halogens under pressure*
2021-07-30MinWu吴旻YeFengWu吴烨峰andYiMa马毅
Min Wu(吴旻) Ye-Feng Wu(吴烨峰) and Yi Ma(马毅)
1College of Materials Science and Engineering,Zhejiang University of Technology,Hangzhou,310014,China
2College of Mechanical Engineering,Zhejiang University of Technology,Hangzhou 310014,China
Keywords: high pressure,structural transition,halogen,first-principles calculation
1. Introduction
The study of the structures and properties of simple diatomic molecular solids under pressure has been one of the most important subjects in highpressure science.[1-14]Various diatomic molecular solids, such as hydrogen,[2,5]nitrogen,[4]and the halogens[3,6-14]undergo molecular-tomonatomic phase transitions,[15]while molecular dissociation usually accompanies insulator-to-metal transitions. When diatomic-molecule solids are compressed, the intermolecular distances decrease dramatically, and the intramolecular distances increase slightly or remain constant. These two distances become comparable under high pressure, leading to molecular dissociation. Meanwhile, under high pressure, delocalized covalent bonds give rise to insulator-to-metal transitions. Experiment also has shown that the molecular states of halogens persist to high pressures beyond gap-closure pressures, indicating that metallization is not directly related to molecular dissociation.[16]The intermediate phase V with incommensurate structures was observed before molecular dissociation took place in the three halogens (Cl2, Br2, and I2),and there were suggestions that it was formed by modulation of the dissociated monatomic phases.[15,17,18]The dissociation pressures of iodine,bromine and chlorine have been suggested to be about 30 GPa, 80 GPa and 250 GPa, respectively.[18,19]The pressuredependent structural evolutions of these three halogens have been suggested to scale well with the function for a reduced atomic volume,defined asVr=Vat/(8r3s),wherersis the interatomic distance of the diatomic molecule.[16]For instance, the semimetallization of iodine at 16 GPa and that of bromine at 60 GPa both take place atVr= 1.37.[20]On the other hand,the molecular dissociation pressure in chlorine was suggested to be 220±40 GPa by the scaling rules,[16]and was eventually confirmed to be~256 GPa in a recent experiment.[18]From the abovementioned highpressure studies on the halogens, it can be seen that most of the research effort has been focused on the dissociation transition from the molecular phase to the monatomic phase. However, a new structural transition with anomalous bondlength behavior has been found in solid bromine under high pressure,[20]which has been suggested to be caused by the increased secondary nearest intermolecular interaction in the later theoretical simulation.[1]However,the anomalous bondlength behavior has not been observed in iodine and chlorine at high pressure,but is worth investigating to achieve a systematic understanding of the solid halogens or general diatomic molecular solids under pressure.
In this study,first-principles calculations were performed to investigate the structural evolutions of iodine and chlorine under pressure. Similar pressureinduced anomalous bondlength behavior to that initially observed in bromine was also predicted for both iodine and chlorine. The calculated transition pressures for iodine and chlorine were~3 GPa and 10 GPa,respectively. Taking account of the previously calculated transition pressure of~7 GPa for bromine,[1]it is apparent that the pressure required for the new structural transition is larger for halogens with smaller atomic numbers. This order is consistent with those of the metallization and dissociation transitions in these three halogens. This study presents insights into the structural evolutions of the halogens under pressure and may be extended to general diatomic molecular solids.
2. Computational method
Density functional theory (DFT)-based first-principles calculations were performed to investigate the pressureinduced structural evolutions of iodine and chlorine. The Viennaab-initiosimulation package (VASP),[21,22]with projector-augmented wave(PAW)potentials[23]was employed for the calculations of geometry optimization and Raman spectra. The Perdew-Burke-Ernzerhof (PBE)[24]generalized gradient approximation (GGA) was used to describe the exchange-correlation functions. The three halogens(Cl2,Br2,and I2) at ambient pressure have the isostructural molecular phase I with the space groupCmca, as shown in Fig. 1.The unit cell employed contained 8 I(Cl)atoms,in which the diatomic molecules were arranged in zigzag patterns in the layers of thebcplane, and the layers were stacked along the crystallographicaaxes. Valence configurations of 3s23p5and 5s25p5were used for the chlorine and iodine atoms, respectively. A plane-wave energy cutoff of 600 eV was used. The Brillouin zone was sampled using a 12×16×8k-point mesh.Convergence test calculations were performed on both the cutoff energies of the plane wave and the k-point mesh, using a convergence criterion of 1 meV/atom for the total energies of the structures. In the geometry optimization calculations, the lattice vectors and the atomic coordinates were relaxed until the Hellmann-Feynman forces on the atoms were less than 0.005 eV/˚A.Moreover,for the calculations of molecular structures such as iodine and chlorine in this study,a correction of the van der Waals interaction is necessary to improve the geometry optimizations and obtain more realistic structures.In the previous study,[1]various methods were used for the van der Waals correction in the calculations of bromine under pressure,and the results calculated using the Grimme-D2 method[25]were suggested to agree better with experimental values. In order to conduct a systematic comparison between these three halogens, the Grimme-D2 method was employed in the present study.

Fig. 1. The Cmca structure of iodine and chlorine under ambient pressure.The black spheres are atoms in the first layer and the white spheres are atoms in the second layer. Here, r1, r2, and r3 represent the intramolecular bond length, the first neighbor intermolecular distance, and the secondary neighbor intermolecular distance,respectively.
3. Results and discussion
3.1. Equations of state
The equations of state (EOS) of theCmcastructures of iodine and chlorine calculated with the Grimme-D2 method are shown in Fig.2 together with a comparison to the experimental results.[16]The comparison shows that the calculation cannot precisely reproduce the experimental results,even with van der Waals corrections. In particular, the volumes at low pressures are underestimated,similarly to the previous calculation for bromine, in which the calculated volume was 6%smaller than the experimental value at ambient pressure.[1]For iodine, the volume of the unit cell is 307.5 ˚A3, which is about 10% smaller than the experimental value at ambient pressure, while the agreement is much better at pressures above 2 GPa. On the other hand, the calculations for chlorine agree much better with the experimental results in the current pressure range from 0 to 40 GPa. At ambient pressure, the volume of the unit cell of chlorine is 224.4 ˚A3,which is only about 2% smaller than the experimental value.The underestimated volume at ambient pressure can be interpreted as overestimated intermolecular interactions. Obviously,it is a systematic error caused by the calculation method,which fails to accurately describe the van der Waals interactions in halogen molecular solids. Figure 3 shows the calculated lattice parameters of iodine and chlorine, compared with the available experimental results.[16]The calculated lattice parameters of iodine at ambient pressure area=7.03 ˚A,b=4.52 ˚A, andc=9.68 ˚A, compared to the experimental values ofa=7.27 ˚A,b=4.79 ˚A, andc=9.80 ˚A,[16]while for chlorine,the calculated lattice parameters at ambient pressure area=6.41 ˚A,b=4.25 ˚A, andc=8.24 ˚A, compared to the experimental values ofa=6.26 ˚A,b=4.51 ˚A, andc=8.32 ˚A.[16]The results show that the lengths of thecaxes of both iodine and chlorine can be accurately reproduced by the calculations, while the calculated lengths of thebaxes in both cases are obviously shorter than the experimental values.This is similar to the previous calculation for bromine.[1]From the abovementioned results,the systematically underestimated volumes at ambient pressure are mainly due to the overestimated intermolecular interaction along thebaxes in thebcplane.
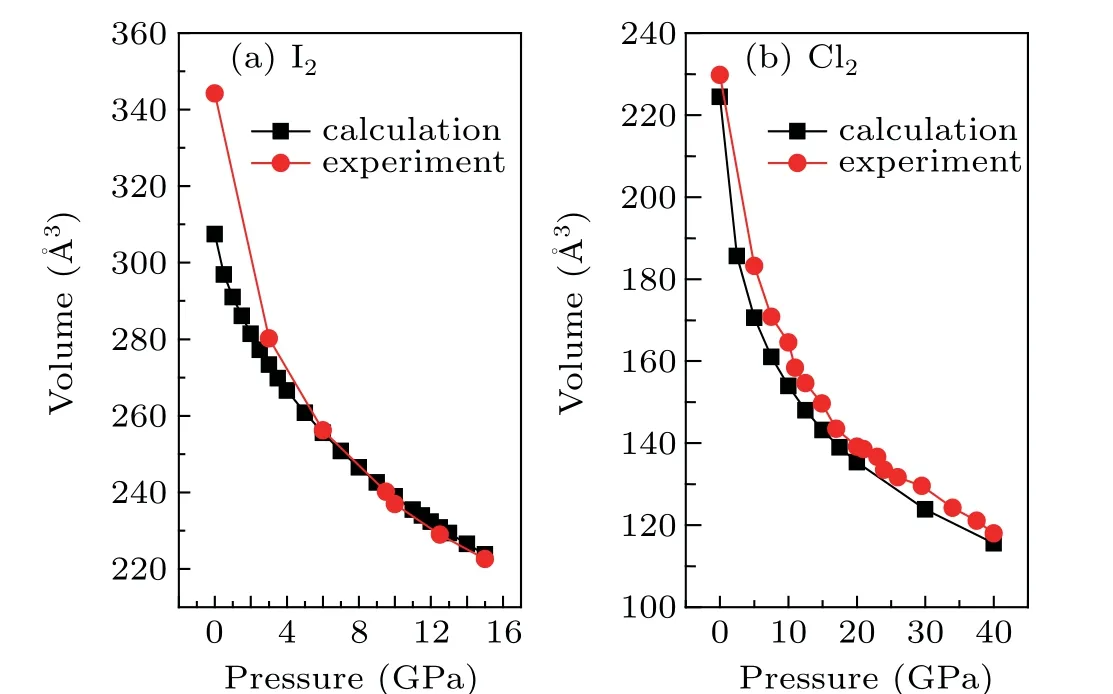
Fig.2. Equations of state comparison between the calculated and experimental results for(a)iodine,(b)chlorine. The solid lines are for visual guidance.The experimental results were obtained from Ref.[16].

Fig.3. The comparison of the lattice parameters between the calculation and experiment results: (a) iodine, (b) chlorine. The solid lines are guides for eyes. The experiment results were obtained from the Ref.[16].
3.2. Raman frequency


Fig. 4. Pressure-dependent Raman vibrational frequencies of (a) iodine,(b) chlorine. (c) The four Raman active modes in iodine and chlorine. The solid lines are visual guides. The experimental results were obtained from Ref. [15], and Li et al.’s theoretical results for chlorine were obtained from Ref.[17].
3.3. Structural analysis
Figure 5 shows the calculated interatomic distances for iodine and chlorine under pressure,wherer1,r2,andr3represent the intramolecular distance (i.e., the I-I and Cl-Cl bond lengths),the first nearest intermolecular distance and the second nearest intermolecular distance, respectively. The calculatedr1of iodine at ambient pressure is 2.805 ˚A,~3%longer than the experimental value of 2.715 ˚A,[16]while for chlorine,the calculatedr1at ambient pressure is 2.01 ˚A,~1.5%longer than the experimental value of 1.98 ˚A.[16]As a reference,the calculatedr1of bromine at ambient pressure is 2.387 ˚A,which is~5% longer than the experimental value of 2.27 ˚A.[16]The systematically elongated intramolecular distancesr1are consistent with the underestimated volumes at ambient pressure. As mentioned above, the underestimated volumes are due to the overestimated intermolecular interactions, which subsequently weaken the intramolecular interaction and result in elongated bond lengths. Thus, these calculated halogen molecular solid structures at ambient pressure act like precompressed structures. As a result, the calculated pressures of the structural transitions are expected to be smaller than the experimental values. For instance, the molecular dissociation pressure of bromine calculated in the previous study using the Grimme-D2 method is~45 GPa[1]which is much smaller than the experimental value of~84 GPa.[15]The previously calculated anomalous bondlength transition pressure of bromine is~7 GPa,which is also much smaller than the experimental value of~25 GPa.[20]In the other theoretical study of chlorine under pressure,[17]the molecular dissociation pressure was predicted to be~157 GPa,while the molecular dissociation pressure was later confirmed to be~256 GPa by experiment.[18]Most importantly, the anomalous bondlength evolution under pressure that has been observed in bromine was also found in both iodine and chlorine in the present calculations. For iodine,the bond lengthr1initially increases from 2.805 ˚A at ambient pressure to 2.807 ˚A at 3 GPa upon compression.At higher pressures,the compression effect becomes dominant and the bond length starts to shrink.For chlorine,the bond lengthr1initially increases from 2.01 ˚A at ambient pressure to 2.026 ˚A at 10 GPa. The transition pressures of these anomalous bondlength evolutions for chlorine, bromine, and iodine are 3 GPa,7 GPa,and 10 GPa,respectively. As a reference, solid hydrogen in the highpressure phase with a space group ofC2/cwas also predicted to have such anomalous bondlength evolution under pressure, and the transition pressure was found to be~300 GPa.[27]This indicates that anomalous bond length evolution under pressure could be general in diatomic molecular solids. From the results presented, it can also be deduced that the influence of the pressure-induced enhanced intermolecular interaction on the bondlength elongation is weaker for stronger covalent bonds such as the Cl-Cl bond, so a higher pressure is needed for the structural transition, and vice versa. Although the transition pressures might be significantly underestimated and fail to follow the scaling rules with the function of the reduced volume, the order of the transition pressures is in good agreement with those of the metallization and molecular dissociation of the halogens under compression.

Fig. 5. Calculated pressuredependent interatomic distances for (a) iodine,(b)chlorine.
The magnitude of the bond-length fluctuation during this new structural transition in halogens under pressure is so subtle that no obvious Raman softening can be found in both the experimental and theoretical Raman spectra.[1,15]However,these subtle changes in the bond lengths are strongly correlated with the intermolecular interaction. Figure 5 shows that the second nearest intermolecular distancer3decreases much faster than the first nearest intermolecular distancer2. For instance, the value ofr3in iodine decreases by 5.3%, from 3.827 ˚A at ambient pressure to 3.627 ˚A at 3 GPa, while the value ofr2only decreases by 3.6%from 3.353 ˚A to 3.232 ˚A.At transition pressures of 3 GPa for iodine and 10 GPa for chlorine,the values ofr3are comparable to those ofr2at ambient pressure. This indicates that the second nearest intermolecular interaction, which was very weak at ambient pressure,becomes unavoidable and could have a significant influence on the structure. Apart from the pressure-induced subtle changes in the bond lengths, the diatomic halogen molecules have more freedom to rotate,and the rotations should be much easier to observe. Figure 6 shows the pressure-dependent intermolecular angles, as illustrated in Fig. 1. Clear transitions at 3 GPa in the results of iodine and at 10 GPa in the results of chlorine can be found in the curve of intermolecular angleθ4.These observations are also similar to the previous results for bromine.[1]This suggests that the structural transitions with the anomalous bond-length evolutions in the three halogens(Cl2, Br2, and I2) are due to the pressureenhanced secondary nearest intermolecular interactions. The enhanced secondary nearest intermolecular interactions are also confirmed by the charge density analyses shown in Fig.7. At ambient pressure,the secondary nearest intermolecular interaction is very weak compared to the first nearest intermolecular interaction.At the transition pressures, it is obvious that the secondary nearest intermolecular interactions have become much stronger and cannot be neglected.

Fig.6.(a)The calculated pressuredependent angular evolutions of(a)iodine,(b)chlorine. The solid lines are visual guides.

Fig. 7. 2D charge density plots in the bc plane for (a) iodine (b) chlorine.The unit of charge density is e/cell. The emergence of the secondary nearest intermolecular interaction is highlighted with a white ellipse.
4. Conclusion and perspectives
In summary, the structural evolutions of two diatomic molecular solids (iodine and chlorine) under pressure were investigated using first-principles calculations. Similarly to the previous theoretical results for bromine, the new structural transitions with anomalous bondlength evolutions were also found in iodine and chlorine under pressure. The bond lengths initially increased with pressure and then started to decrease above the transition pressures. The calculated pressures for this new structural transition were 3 GPa, 7 GPa,and 10 GPa for iodine, bromine, and chlorine, respectively.These results indicated that such pressureinduced anomalous bondlength evolutions could be general in halogens, or even other diatomic molecular solids. Despite the underestimated values of the transition pressures, which were mainly due to imperfect van der Waals corrections, the order of the transition pressures was consistent with the other structural transitions in solid halogens under pressure, such as metallizations and molecular dissociations. Further analyses of the intermolecular distances, intermolecular angles, and charge densities showed that the anomalous bondlength evolution is due to an enhanced secondary nearest intermolecular interaction.Upon initial compression,the enhanced secondary nearest intermolecular interaction weakens the intramolecular interactions,thus leading to elongated bonds. With further compression,the compression effects become dominant and the elongated bonds start to shrink. These results will trigger further experiments to confirm the predicted structural transitions in iodine and chlorine,which will improve our understanding of the general structural behavior of diatomic molecular solids under pressure,including hydrogen.
杂志排行

Chinese Physics B的其它文章
- Numerical simulations of partial elements excitation for hemispherical high-intensity focused ultrasound phased transducer*
- Magnetic-resonance image segmentation based on improved variable weight multi-resolution Markov random field in undecimated complex wavelet domain*
- Structure-based simulations complemented by conventional all-atom simulations to provide new insights into the folding dynamics of human telomeric G-quadruplex*
- Dual-wavelength ultraviolet photodetector based on vertical(Al,Ga)N nanowires and graphene*
- Phase-and spin-dependent manipulation of leakage of Majorana mode into double quantum dot*
- Deep-ultraviolet and visible dual-band photodetectors by integrating Chlorin e6 with Ga2O3