HONO在γ-Al2O3 (110)表面非均相氧化SO2机理的第一性原理研究
2021-07-29林艳辉宋绵新李海龙罗伟格张金梅罗伟恢
林艳辉,边 亮,宋绵新, ,李海龙,李 宇,罗伟格,张 娇,张金梅,罗伟恢,张 琴
(1. 西南科技大学 材料科学与工程学院, 四川 绵阳 621010; 2. 固体废物处理与资源化教育部重点实验室,四川 绵阳 621010)
硫酸盐作为PM2.5的主要成分,是影响全球气候变化和空气质量的主要因素之一(Liuetal., 2012; Fangetal., 2017; Elizabeth and Marcelo, 2018)。SO2作为硫酸盐气溶胶形成的重要前驱体(Maetal., 2008),可以通过大气中的氧化剂,如羟基自由基(·OH)、臭氧、Criegee自由基等氧化形成硫酸盐(Jayneetal., 1990; Sieveringetal., 1992; Capaldoetal., 1999; Bouyaetal., 2015)。研究者通过场地观测和模型研究表明,在矿物气溶胶颗粒物表面非均相反应形成的硫酸盐对硫酸盐气溶胶的形成起着至关重要的作用(Zhaoetal., 2018)。因此,研究SO2在矿物表面的非均相转化机理对硫酸盐的形成具有重要意义。密度泛函理论(density functional theory, DFT)通常作为重要的研究手段用来研究SO2在矿物表面的吸附机制与非均相反应机理。例如,Kaewruksa等(2013)利用DFT研究了SO2在ZnO矿物表面的吸附机制,其主要是通过与表面活性位点(Zn原子和氧缺陷)形成较强共价键而吸附(吸附能-35.06 kcal/mol)在矿物表面(Tangetal., 2015)。
氧化铝(Al2O3)是矿物粉尘的主要成分之一,质量浓度为15%(Lianetal., 2019)。γ-Al2O3由于具有较高的比表面积、较好的化学稳定性和热稳定性,常被应用于研究矿物与大气痕量气体的非均相反应,进而解释非均相反应机制(Genetal., 2019)。例如,Li等(2020)通过DFT模拟了H2O2在γ-Al2O3(110)表面非均相氧化SO2的机制。研究证明H2O2是通过在矿物氧化物表面分解产生·OH氧化SO2形成硫酸盐的(李冬坤等, 2019)。同时,Harris等(2013)利用高精度同位素比值质谱法证实了SO2在矿物表面被·OH氧化的过程,这为探讨·OH在矿物表面非均相氧化SO2反应提供了重要参考。亚硝酸(HONO)作为一种重要的中间产物和大气物种,是白天·OH产生的重要来源(贡献约30%~60%),不仅影响许多化学活性气体的大气化学反应过程而且影响二次气溶胶的形成(Zeinetal., 2013; Maetal., 2017; Chenetal., 2019; Yangetal., 2020)。然而,目前关于HONO在矿物表面氧化SO2的非均相反应机理和HONO在非均相反应过程中的作用的相关报道较少。因此,研究HONO在γ-Al2O3(110)上的分解过程和HONO对SO2的非均相氧化机理,对揭示HONO在非均相反应过程中的作用和矿物表面的非均相氧化机理具有重要的意义。
考虑到·OH的不稳定和瞬时性等因素难以在实验中直接检测,因此,为了阐明SO2在γ-Al2O3(110)表面上被·OH氧化的机理,本文采用DFT计算了SO2、HONO在γ-Al2O3(110)表面的吸附机制,进一步分析了HONO在完整或缺陷γ-Al2O3(110)表面上非均相氧化SO2的机制。研究结果不仅为理解HONO在矿物表面非均相氧化SO2提供了理论基础,而且为SO2+HONO在γ-Al2O3(110)表面进行催化反应提供了新技术。
1 计算细节
1.1 计算模型

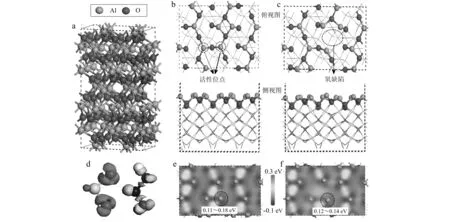
图 1 γ-Al2O3 (110)表面的结构和差分电荷密度Fig. 1 The structure and charge density difference of γ-Al2O3 (110) surfacea—γ-Al2O3的晶体结构; b, c—完整的和氧缺陷的γ-Al2O3 (110)表面的俯视图和侧视图; d—SO2和HONO的差分电荷密度; e, f—完整的和氧缺陷的γ-Al2O3 (110)表面的差分电荷密度 a—the crystal structure of γ-Al2O3; b, c—top and side views of the perfect and the oxygen defect surface of γ-Al2O3 (110); d—the charge density difference of SO2 and HONO; e,f—charge density difference between the perfect and the oxygen defect surface of γ-Al2O3 (110)
1.2 计算方法
本文计算基于量子力学中的DFT方法,使用Materials Studio的Dmol3软件包,选择了基于广义梯度近似下的Perdew-Burke-Ernzerhof(PBE)泛函。由于Dmol3软件包对周期结构的计算效率较高,因此被用来优化初始结构、电子结构的计算和搜索过渡态(transition states, TS)。用有效核势(effective core potential,ECP)和DND(double numerical plus d-functions)基组描述体系的电子,最后利用Dmol3计算体系的电子结构和电子转移机制。其中,用3×3×3的Monkhorst-Pack k点计算布里渊区积分。收敛阈值和SCF(self consistent field)收敛精度分别设置为10-6Ha和10-5Ha。
吸附能(E, eV)定义为:E=ET-EA-ES, 式中,ET是γ-Al2O3(110)表面吸附后的总能量,EA和ES分别为吸附前吸附物的能量和表面的能量。
2 结果与讨论
2.1 SO2在γ-Al2O3 (110)表面的吸附
为了研究SO2在γ-Al2O3(110)表面上的吸附,不仅考虑了SO2的初始位置(水平和垂直),而且还考虑了γ-Al2O3(110)表面上的吸附位点(Al和O)。通过结构优化得到的吸附构型主要有2种: 1A和1B构型分别是由SO2中的O和S原子与Al2O3中Al原子结合,2A和2B构型是在氧缺陷表面上的吸附构型,如图2a所示。对比表1不同吸附构型的吸附能发现,不论在完整γ-Al2O3(110)表面还是在氧缺陷表面,吸附构型1A(-0.12 eV)和2A(-2.06 eV)的吸附能均小于1B(0.46 eV)和2B(-0.15 eV),这说明SO2在γ-Al2O3(110)表面的化学吸附是通过SO2的O原子吸附在γ-Al2O3(110)表面。同时,吸附构型2A的吸附能更低,因此含氧缺陷的表面更有利于SO2在γ-Al2O3表面的化学吸附。相应的Al—O键为1.86 Å。

表 1 不同吸附构型的吸附能 eVTable 1 The adsorption energy of different adsorption configurations
为了进一步分析SO2在γ-Al2O3(110)表面上的吸附机制,本文分析了吸附构型1A和2A的O原子和表面Al原子的局域态密度(PDOS),如图2所示。在吸附构型1A中Al-3p和O-2p轨道在能量区域(-4.7~-1.5 eV)和(-9.4~-6.0 eV)有明显重叠,这形成了p-p(Al-3p-O-2p)杂化轨道。在吸附构型2A中,Al-3p和O-2p轨道在能量区域(-5.0~-1.5 eV)和(-9.0~-6.0 eV)有明显重叠,重叠程度显著高于1A。因此, 2A构型的吸附能较低。Mulliken电荷布局分析作为定量分析电子转移的重要依据,由表2所示,完整γ-Al2O3(110)表面Al和O的电荷分别在吸附过程中升高,SO2表面S和O的电荷明显减少。这说明Al和S(或O)原子的Mulliken电荷在吸附过程中电子转移是从γ-Al2O3(110)表面向SO2进行,吸附构型1A和2A电子转移分别为0.20 e和0.23 e。这也说明在含氧缺陷的γ-Al2O3(110)表面的吸附构型2A相互作用强于1A。因此,γ-Al2O3(110)表面的Al作为SO2的活性位点,吸附构型2A作为最优吸附方式,p-p(Al-3p-O-2p)轨道杂化是Al—O键形成的主要原因。
2.2 HONO在γ-Al2O3 (110)表面的吸附
HONO在矿物上发生非均相反应产生具有氧化性的·OH,为SO2的氧化提供了重要途径(Elshorbanyetal., 2008)。由于·OH的存在更有利于矿物的非均相反应发生,因此,有必要研究HONO在γ-Al2O3(110)表面的吸附机制。考虑到不同吸附位点,得到了HONO在γ-Al2O3(110)表面上的吸附构型,如图3a所示。3A和3B分别是HONO中ON和N与Al2O3中Al原子在完整表面上的吸附构型,4A、4B是HONO中OH、ON和N与Al原子在缺陷表面上的吸附构型。由表1可知,HONO在完整γ-Al2O3(110)表面吸附时3B(-1.05 eV)的吸附能低于3A(0.92 eV),在含氧缺陷γ-Al2O3(110)表面吸附时4B(-3.53 eV)的吸附能低于4A(1.29 eV)。进一步对比发现4B的吸附能最低,这说明HONO在γ-Al2O3(110)表面的吸附是通过HONO分解后以N和OH与Al2O3中的Al原子发生的,同时在含氧缺陷的γ-Al2O3(110)表面更有利于HONO的吸附。
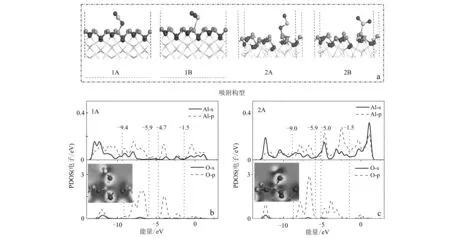
图 2 SO2在γ-Al2O3 (110)表面的吸附情况Fig. 2 Adsorption details of SO2 on γ-Al2O3 (110) surfacea—SO2在γ-Al2O3 (110)表面的吸附构型; b、c—吸附构型1A和2A的O与表面Al原子的PDOS分析a—the structure of SO2 adsorption on γ-Al2O3 (110) surface; b, c—PDOS analysis of O and Al atoms in adsorption configurations 1A and 2A

图 3 HONO在γ-Al2O3 (110)表面的吸附情况Fig. 3 Adsorption details of HONO on γ-Al2O3 (110) surfacea—HONO在γ-Al2O3 (110)表面的吸附构型; b—吸附构型3A和4A的ON、OH和表面Al原子的PDOS分析; c—吸附构型4B的OH+N、表面Al原子的PDOS分析a—the structure of HONO adsorbed on γ-Al2O3 (110) surface; b—PDOS analyses of ON, OH, and Al atoms in adsorption configurations 3A and 4A; c—PDOS analyses of OH+N and Al atoms in adsorption configuration 4B
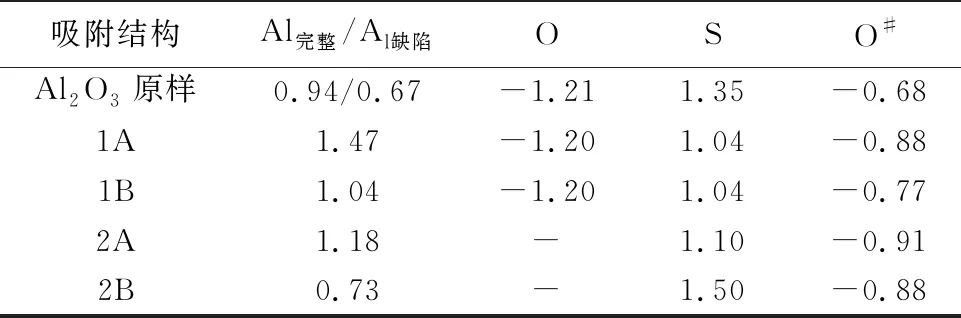
表 2 SO2在γ-Al2O3 (110)表面不同吸附构型的Mulliken电荷eTable 2 Mulliken charge of SO2 on the surface of γ-Al2O3 (110) with different adsorption configurations
为了进一步描述HONO在γ-Al2O3(110)表面的吸附机制,对吸附构型3A、4A和4B的ON、OH和OH+N及表面Al原子进行PDOS分析,如图3b、3c所示。图3b中Al-3p和O-2p轨道在能量区域-0.9~1.6 eV和-6.8~-5.1 eV有明显重叠,形成了p-p(Al-3p-O-2p)轨道杂化。图3c中吸附构型4B的Al-3p、O-2p和N-2p轨道在能量区域(-2.1~1.3 eV)、(-8.4~-3.2 eV)有明显重叠,形成了p-p(Al-3p-O-2p-N-2p)轨道杂化。吸附构型4B的轨道重叠程度和局域高度显著高于3A和4A,因此HONO在γ-Al2O3(110)表面上的吸附更倾向于p-p(Al-3p-O-2p-N-2p)轨道杂化。通过Mulliken电荷分析发现, HONO在γ-Al2O3(110)表面吸附时Al和O的电荷增大,OH和N、ON电荷减小,这证明Al和N(或O)原子的Mulliken电荷在吸附过程中电子转移是从γ-Al2O3(110)表面向HONO转移。通过PDOS和Mulliken电荷布局分析可以发现HONO的分解遵循Haber-Weiss机制(Songetal., 2017),即电子从表面Al原子转移到HONO分子,生成·OH和NO。吸附构型3A、4A和4B的电子转移分别为0.01 e、0.03 e和0.54 e,这说明吸附构型4B的相互作用强于3A和4A。因此,4B作为HONO在γ-Al2O3(110)表面的最优吸附方式,主要是以N和OH与γ-Al2O3(110)表面的Al结合,p-p(Al-3p-O-2p-N-2p)轨道杂化是成键的主要原因。

表 3 HONO在γ-Al2O3 (110)表面不同吸附构型的Mulliken电荷eTable 3 Mulliken charge of HONO on the surface of γ-Al2O3 (110) with different adsorption configurations
2.3 SO2和HONO在γ-Al2O3 (110)表面共同吸附
仅仅计算HONO在γ-Al2O3(110)表面不能直接证实·OH氧化性的存在,因此又计算了SO2和HONO在γ-Al2O3(110)表面共同吸附的作用。考虑到SO2和HONO的竞争吸附以及HONO的分解,分别得到了5种吸附构型,如图4a所示。5A和5B分别是SO2中的O和HONO中的N与Al原子在完整γ-Al2O3(110)表面上的吸附构型,6A和6B是SO2中的O和HONO中的N与Al原子在缺陷表面上的吸附构型,6C是HONO分解成NO和·OH与SO2共同吸附在缺陷表面上的吸附构型。由表1可知,5B(-0.88 eV)和6B(-2.15 eV)的吸附能均小于5A(-0.26 eV)和6A(-1.93 eV)的吸附能,这说明SO2和HONO的吸附之间存在竞争性吸附,相比较而言HONO比SO2优先吸附在γ-Al2O3(110)表面。吸附构型6C中,HONO在含氧缺陷的γ-Al2O3(110)表面分解形成·OH和NO,·OH迅速与SO2反应形成HOSO2团簇分子。5种吸附构型中6C(-2.87 eV)的吸附能最低,因此含氧缺陷的γ-Al2O3(110)表面更有利于SO2和HONO的共同吸附。

图 4 SO2+HONO共吸在γ-Al2O3 (110)表面的吸附情况Fig. 4 Co-adsorption details of SO2 + HONO on γ-Al2O3 (110) surfacea—SO2+HONO共吸在γ-Al2O3 (110)表面的结构; b—吸附构型6C的N+OH和表面Al原子的PDOS图a—the structure of SO2 + HONO co-adsorption on γ-Al2O3 (110); b—PDOS analyses of N+OH and Al atoms in adsorption configuration 6C
为了进一步研究SO2和HONO在γ-Al2O3(110)表面的共同吸附机制和·OH对SO2氧化作用,分析了吸附构型6C吸附的SO2、N和OH及表面Al原子的PDOS分布,如图4b所示。Al-3p和N-2p轨道在(-0.36~1.2 eV)和(-9.7~-7.3 eV)有显著重叠,形成了p-p(Al-3p-N-2p)轨道杂化。SO2中的O-2p和·OH中的O-2p在(-9.3~ 0.9 eV)有明显重叠,形成了p-p(O-2p-O-2p)轨道杂化。因此,SO2和HONO的共同吸附更倾向于p-p(Al-3p-N-2p)和p-p(OS-2p-OH-2p)轨道杂化。同时,由于氧缺陷存在增强了p-p轨道杂化,因此6C具有较低的吸附能。通过对比表4中不同构型的Mulliken电荷发现,HONO在γ-Al2O3(110)表面上吸附时,电子转移是从γ-Al2O3(110)表面向HONO分解产生的NO转移,由SO2向·OH转移。吸附构型5A、5B、6A、6B和6C电子转移分别为0.18 e、0.15 e、0.22 e、0.2 e、和0.41 e,可见吸附构型6C的相互作用最强。因此,吸附构型6C作为SO2和HONO在γ-Al2O3(110)表面吸附的最优方式, p-p(Al-3p-N-2p)和p-p(OS-2p-OH-2p)轨道杂化是主要的原因。

表 4 SO2+HONO在γ-Al2O3 (110)表面不同吸附构型的Mulliken电荷eTable 4 Mulliken charges of SO2 + HONO on the surface of γ-Al2O3 (110) with different adsorption configurations
3 结论
本文重点研究了SO2和HONO在γ-Al2O3(110)上的非均相氧化机理,计算了SO2和HONO在γ-Al2O3(110)表面上的吸附机制。计算表明,SO2在γ-Al2O3(110)表面主要以分子形式吸附。2A作为最优吸附方式,p-p(Al-3p-O-2p)轨道杂化是Al—O键形成的主要原因。HONO在含氧缺陷γ-Al2O3(110)表面上吸附以分解形式通过N和OH与Al结合,形成的p-p(Al-3p-O-2p-N-2p)轨道杂化是成键的主要原因。SO2和HONO共同吸附在γ-Al2O3(110)表面时,HONO优先吸附在γ-Al2O3(110)表面,分解产生的·OH与SO2发生氧化作用形成HOSO2团簇分子在γ-Al2O3(110)表面发生吸附作用。吸附过程中主要以OS、HONO中的OH和N与γ-Al2O3(110)表面Al原子结合,形成的p-p(OS-2p-OH-2p;Al-3p-N-2p)轨道杂化是成键的主要原因。本研究为理解HONO在矿物表面氧化SO2的非均相氧化机理提供了理论基础并为硫酸盐气溶胶形成提供了参考。
