乌鳢水泡病毒L蛋白的原核表达及多克隆抗体制备
2021-07-26张艳维张永安涂加钢
张艳维,张永安,涂加钢
华中农业大学水产学院/农业微生物学国家重点实验室,武汉430070
弹状病毒是与人类疾病及工业、农业密切相关的一种重要病毒,其宿主范围广泛,主要包括哺乳动物、鸟类、昆虫、鱼类、爬行动物及植物等[1],导致宿主患病或死亡,造成重大的经济损失。弹状病毒是一类不分节段的单股负链RNA病毒,具有包膜,病毒粒子长100~430 nm,直径45~100 nm[2]。目前已经分离出来的鱼类弹状病毒有十几种[3]。乌鳢水泡病毒(snakehead vesiculovirus,SHVV)是仲恺农业工程学院林蠡教授在2014年从广东顺德市的一个乌鳢养殖场中分离得到的1株弹状病毒,其基因组全长约11 kb,主要编码5种结构蛋白:核蛋白(N)、磷蛋白(P)、基质蛋白(M)、糖蛋白(G)和RNA依赖性RNA聚合酶蛋白(L),属于弹状病毒科(Rhabdoviridae)、Perhabdovirus属。感染SHVV的鱼会出现典型的弹状病毒病症状,主要包括出血、水肿等[4]。
L蛋白是弹状病毒的一种结构蛋白,同时也是分子质量最大的弹状病毒蛋白,约240 ku,具有RNA聚合、mRNA封端和封端甲基化等功能[5-7]。据报道β微管蛋白、延伸因子1和鸟苷酸转移酶均与水泡性口炎病毒(vesicular stomatitis virus,VSV)的L蛋白相互作用,并合成VSV RNA的必要因子[8-10]。DNA 拓扑异构酶1与埃博拉病毒L蛋白相互作用,是L蛋白聚合酶活性所必需的[11]。动力蛋白轻链 1作为转录因子,通过与病毒L蛋白结合促进狂犬病毒的初级转录[12]。虽然L蛋白的功能在人类或其他哺乳动物弹状病毒感染中已得到广泛研究[11-13],但其在鱼类弹状病毒感染中的作用尚无报道。
由于SHVV是一种新的鱼类弹状病毒,目前还没有有效的防治SHVV感染的措施。基于L蛋白在病毒转录和复制中的重要作用,本研究将L基因前900个碱基构建在原核表达载体pET-32a(+)上,进行表达及纯化,并将纯化的蛋白免疫新西兰大白兔,制备SHVV L蛋白多克隆抗体,以期为后续SHVV的致病机制和L蛋白的功能研究奠定基础。
1 材料与方法
1.1 试验材料来源
SHVV由林蠡教授分离并在笔者所在实验室保存。斑点叉尾鮰卵巢细胞系(CCO)、质粒pET-32a(+)和pCDNA-L由笔者所在实验室保存。试验所用Trans 5ɑ大肠杆菌克隆菌株和BL21大肠杆菌表达菌株均购自北京全式金生物技术有限公司。反转录试剂盒和限制性内切酶购自宝生物工程(大连)有限公司。质粒抽提试剂盒、PCR产物纯化回收、胶回收试剂盒购自广州美基生物科技有限公司。
1.2 引物合成
根据GenBank上发表的序列(登录号:KP876483.1),利用Primer5.0软件设计1对引物,引物序列如下:
L-FW:5′ -TGGGATCCGGTACC
AAGCTTATGGATTATTCTCAGGAATA-3′;
L-BW:5′ -TGGTGGTGGTGGTGCTCGAG
AGATAATTGAAGATTACACA-3′
其中,下划线为酶切位点(HindⅢ和XhoⅠ),送至武汉擎科生物有限公司进行合成。
1.3 pET32a-L重组表达质粒的构建
1)PCR扩增目的基因。以含有SHVVL基因的质粒pCDNA-L为模板,L-FW和L-BW为引物,PCR扩增L基因的前900 bp。扩增程序如下:98 ℃变性10 s、55 ℃退火5 s、72 ℃延伸30 s,30个循环,PCR扩增结束后,产物用1%的琼脂糖凝胶电泳进行检测。
2)酶切、连接和转化。将原核表达载体pET-32a(+)用限制性内切酶HindⅢ和XhoⅠ进行双酶切,1 h后将酶切产物经琼脂糖凝胶电泳分离后使用凝胶回收试剂盒进行回收、纯化。并采用Clone Express One Step Cloning Kit将纯化回收的L片段和酶切后的载体pET-32a(+)进行连接,将连接产物转化至大肠杆菌Trans 5ɑ感受态细胞中。步骤为:冰浴30 min;42 ℃水浴45 s后立刻冰浴2 min;加入400 μL LB液体培养基后放入37 ℃摇床中培养1 h;吸取100 μL菌液均匀涂布在含氨苄的LB固体培养基中;置于37 ℃培养箱培养12 h。
3)重组质粒的提取及鉴定。挑取单克隆菌落接种到含氨苄的LB液体培养基中,将其置于37 ℃的摇床中进行培养,按照质粒抽提试剂盒操作步骤提取重组质粒。以载体pET-32a(+)的通用测序引物(T7-FW 5′-TAATACGACTCACTATAGGG-3′和T7-BW 5′ -GCTAGTTATTGCT CAGCG -3′)对重组质粒进行PCR鉴定,将阳性质粒送至武汉擎科生物有限公司测序。测序结果与GenBank中SHVVL基因序列(登录号:KP876483.1)进行核苷酸序列比较分析。
1.4 重组His-L蛋白的表达和纯化
将测序正确的重组表达质粒转化至BL21(DE3)感受态细胞中,并将PCR鉴定为阳性的菌落接种在含氨苄的LB液体培养基中,置于37 ℃、180 r/min的摇床中震荡培养。当菌液OD值达到0.6时加入IPTG至其终浓度为1 mmol/L。并在16 ℃、150 r/min的摇床中过夜诱导表达。离心收集诱导表达的菌液,并用PBS重悬洗涤2次后进行高压破碎,4 ℃、12 000 r/min离心30 min,收集上清和沉淀,进行SDS-PAGE鉴定分析。
通过上述分析发现重组蛋白表达在上清中,对转化了重组质粒的大肠杆菌进行大量诱导表达,离心后收集菌体,PBS重悬。4 ℃高压破碎,离心收集上清。采用Ni-NTA His-Band 树脂亲和层析柱试剂盒(Qiagen,德国)纯化重组蛋白,参照生工生物Ni-NTA纯化树脂预装柱操作说明进行纯化。再使用蛋白质浓缩管通过离心方法浓缩重组蛋白。浓缩后的重组蛋白通过SDS-PAGE电泳鉴定。
1.5 多克隆抗体的制备
将纯化后的重组His-L蛋白免疫新西兰大白兔,分3次进行免疫,第1次免疫时将蛋白抗原与等量的弗式完全佐剂混合,充分乳化,采用皮下多点注射法免疫新西兰大白兔。第2次和第3次免疫分别在第1次免疫后第2、4周后进行,采用蛋白抗原与等量的弗式不完全佐剂混合,乳化后进行皮下多点注射。在第3次免疫7 d后经心脏取血,4 ℃隔夜静置,5 000 r/min离心30 min,收集血清。检测多克隆抗体的效价,并将血清保存在-80 ℃冰箱备用。
1.6 Western blot 检测
使用T25细胞瓶在含有10%热灭活胎牛血清(Gibco,New Zealand)、青霉素(100 μg/mL)和链霉素(100 μg/mL)的MEM(HyClone,USA)培养基中培养CCO细胞,待细胞长满单层后感染SHVV,并把未感染SHVV的CCO细胞作为对照。24 h后收集细胞样品,加入SDS上样缓冲液,将样品放置在金属浴中煮沸10 min,进行Western blot分析。具体步骤如下:配制6%的聚丙烯酰胺;点样后80 V进行电泳;待溴酚蓝指示剂到达分离胶底部时,结束电泳,并用转膜仪转印装置将蛋白条带电转印到PVDF膜上,以9 V恒压电转印60 min;5%脱脂奶粉封闭2 h,将一抗(L多抗1∶1 000稀释;GST标签抗体1∶5 000稀释)4 ℃过夜孵育,TBST缓冲液清洗3次,每5 min清洗1次;HRP标记的兔抗或者鼠抗作为二抗孵育1 h;TBST缓冲液清洗3次,每次10 min。将PVDF膜置于ECL发光液中,采用化学发光法显色、拍照分析。
1.7 间接免疫荧光检测
将长满单层状况良好的CCO细胞接种到培养皿中,待细胞长至70%~80%时,感染SHVV,并用未感染SHVV的CCO细胞作为对照。孵育1 h后换成5% FBS的MEM培养基继续培养,24 h后吸弃培养基,用PBS洗涤3次,加入4%多聚甲醛室温固定15 min;PBS洗涤3次后,加入0.5% Triton,室温孵育15 min;PBS洗涤3次后,加入5% PBSA封闭1 h;加入L蛋白多克隆抗体,37 ℃孵育2 h;PBS洗涤3次后,加入FITC标记的荧光二抗(1∶100~1∶200),37 ℃避光孵育1 h;PBS洗涤3次后,用DAPI核染15 min,再用PBS洗涤3次。用激光共聚焦显微镜观察并拍照。
2 结果与分析
2.1 SHVV L基因的克隆
用含有SHVVL基因的质粒pCDNA-L为模板进行PCR扩增,产物经1%琼脂糖凝胶电泳分析,可见约为900 bp的特异性条带(图1),与目的片段大小一致。
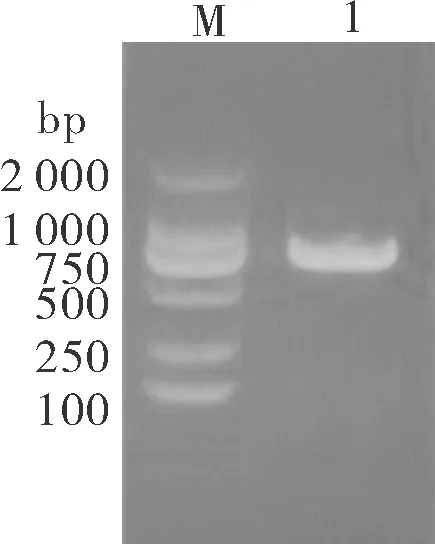
M:DNA marker 2000;1:L基因。M:DNA marker 2000; 1:Target gene of L.
2.2 诱导表达并纯化His-L蛋白
对诱导表达的菌液进行高压破碎,收集上清、流穿液及洗脱下来的蛋白进行SDS-PAGE凝胶电泳分析(图2),结果显示,该融合蛋白在上清中表达,且在第2次洗脱下来的浓度最高,往后依次降低,蛋白分子质量约42 ku,与目的蛋白大小预期一致。收集纯化浓缩后的融合蛋白样品,及诱导前的样品用His标签抗体进行Western blot鉴定,结果显示,诱导前的样品无条带,纯化后的His-L重组蛋白样品在42 ku左右有1条清晰的带,与预期大小一致。
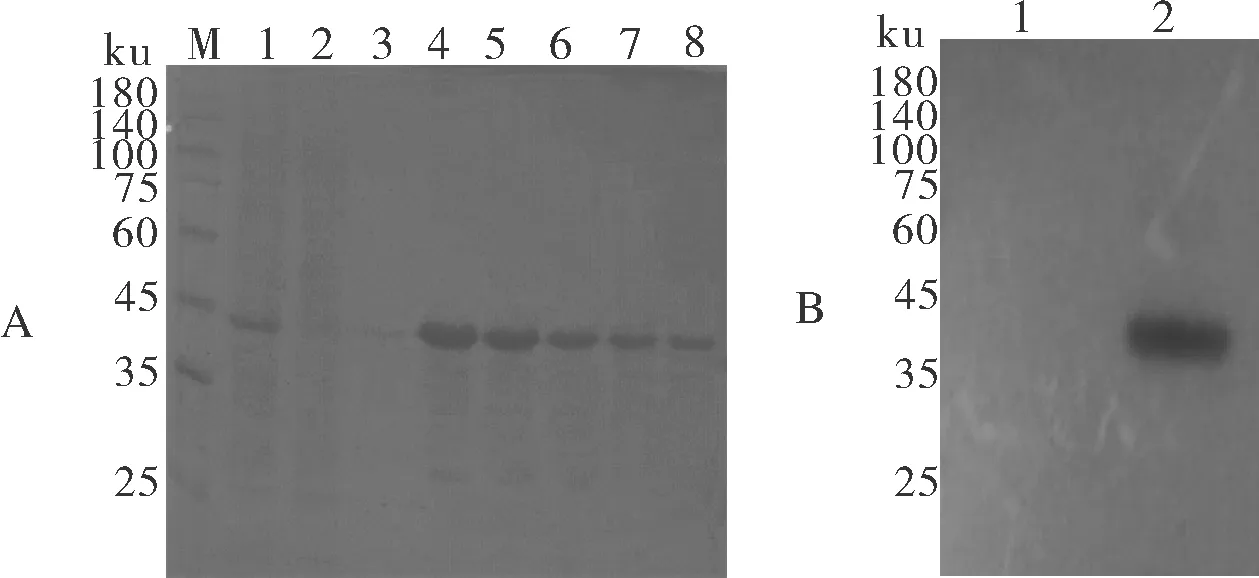
A:SDS-PAGE 鉴定纯化的His-L重组蛋白。 M:蛋白 marker; 1:诱导后上清; 2:流穿液; 3~8:纯化后洗脱的蛋白; B:His-L重组蛋白的鉴定。 1:诱导前蛋白样;2:纯化的His-L重组蛋白样。A:Purified recombinant His-L protein by SDS-PAGE. M:Protein marker; 1:Supernatant of bacteria after induction; 2:Liquid flow-through; 3-8:Purified protein eluted after purification; B:Identification of His-L recombinant protein.1:Protein before induction; 2:Purified recombinant protein His-L.
2.3 Western blot验证L蛋白多抗
重组蛋白His-L经纯化后免疫新西兰大白兔,收集血清获得L蛋白的多克隆抗体。通过Western blot进行分析,结果显示,感染SHVV的蛋白样品有1条单一的条带,大小约220 ku,与目的蛋白大小一致(图3),而未感染SHVV的CCO细胞样品没有出现条带,表明制备的多克隆抗体能特异性识别L蛋白。
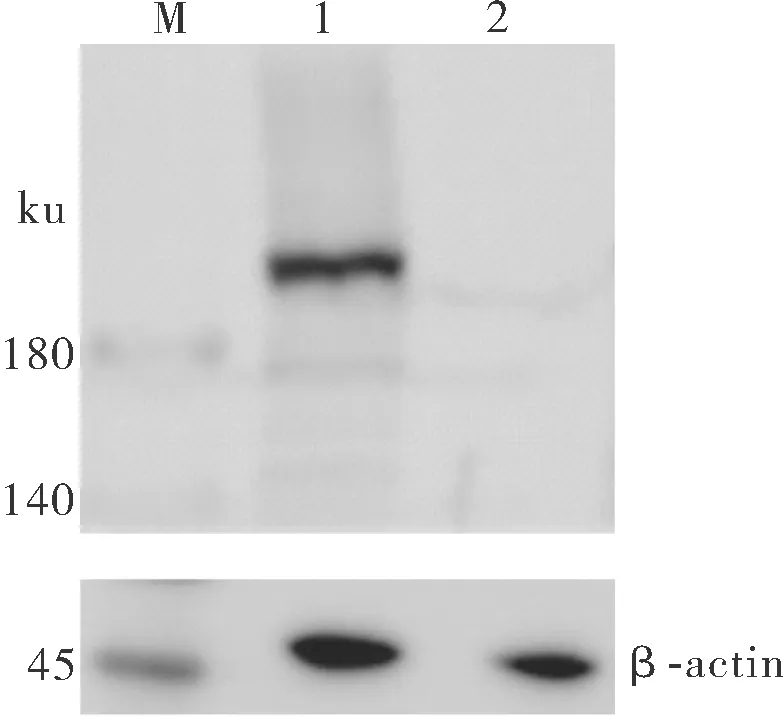
M:蛋白 marker; 1:感染 SHVV的CCO细胞样; 2:未感染SHVV的CCO细胞样。M:Protein marker; 1:SHVV infected CCO cell; 2:Uninfected CCO cell.
2.4 间接免疫荧光实验研究L蛋白的亚细胞定位
将SHVV感染CCO细胞,通过间接免疫荧光观察L蛋白的定位情况。结果显示,L蛋白多克隆抗体可以特异性地识别感染SHVV的CCO细胞中的L蛋白,且L蛋白定位在细胞质中,这与SHVV在细胞质完成基因组的转录、复制一致。而对照组则没有观察到绿色荧光(图4)。表明制备的多克隆抗体可以特异性地与L蛋白结合。
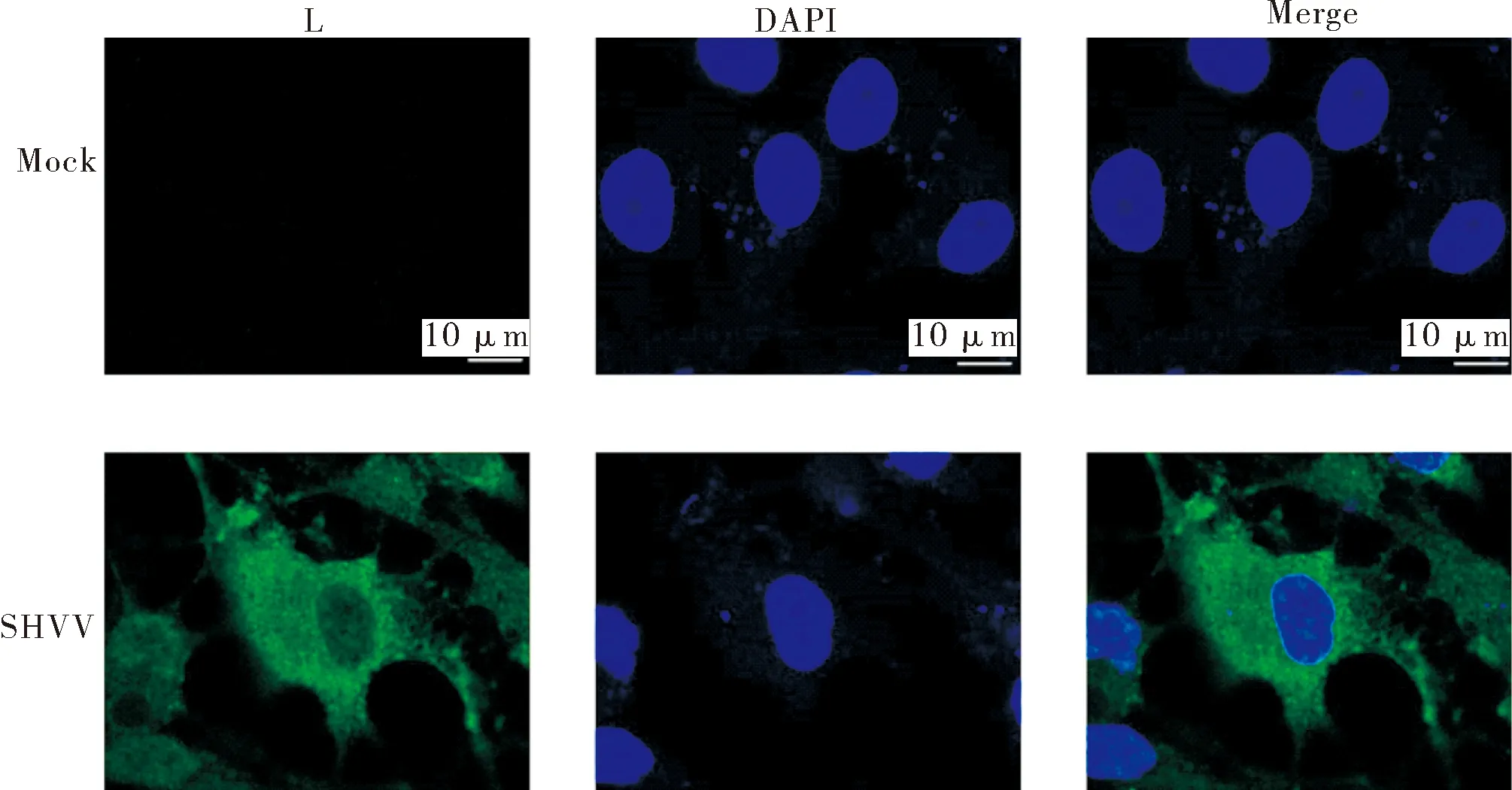
图4 间接免疫荧光分析L多克隆抗体特异性
3 讨 论
乌鳢是我国重要的淡水养殖品种,SHVV是从发病乌鳢中分离得到的1株弹状病毒,SHVV感染给我国乌鳢养殖业造成了巨大的经济损失。弹状病毒的L蛋白有5个功能结构域:RdRp、Cap、CD(连接器域)、MT(甲基转移酶结构域)和CTD(C端结构域)[14],其中RdRp是主要的功能结构域,发挥聚合酶的功能。Rahmeh等[15]的研究发现,VSV P蛋白可以与L蛋白结合,并将L蛋白C端球状结构域(CD、MT和CTD)稳定地锚定在N端RdRp和CAP结构域。Qiu等[16]对VSV的L蛋白N末端结构域进行连续突变,表明N末端许多氨基酸对于病毒转录是必需的。本研究的目的是制备SHVV L蛋白的多克隆抗体,为研究其功能提供基础资料。但因为SHVV L蛋白分子质量大(240 ku),原核表达及纯化较困难,因此,本研究在制备L蛋白多克隆抗体时选择RdRp结构域的前900 bp进行克隆、蛋白表达和纯化以及制备多克隆抗体。这也是本研究的不足之处,后续有待更深入地对RdRp全结构域进行研究,以及进一步开展L蛋白与宿主蛋白互作、L蛋白的翻译后修饰等深入研究。
我们用制备的L蛋白多克隆抗体检测SHVV感染CCO细胞过程中的L蛋白,能观察到1条清晰的目的蛋白条带,说明L蛋白RdRp结构域具有很好的免疫原性。但是也能观察到一些杂带,说明L蛋白多克隆抗体可能与细胞蛋白或其他病毒蛋白发生非特异性结合。
我们利用L蛋白多克隆抗体对SHVV感染CCO细胞过程中的L蛋白的亚细胞进行了定位研究,但是只是对SHVV感染CCO细胞24 h的L蛋白进行亚细胞定位,没有对病毒感染不同时间点开展研究,因而无法确定L蛋白在SHVV感染的早期和晚期是否发生定位的改变,后续有待进一步深入研究。
