牛凸隆病毒HE基因分子特征分析
2021-07-26赵龙,汤承,2,岳华,2*
赵 龙,汤 承,2,岳 华,2*
(1.西南民族大学畜牧兽医学院,成都 610041;2.青藏高原动物遗传资源保护与利用教育部重点实验室,成都 610041)
牛凸隆病毒(bovine torovirus,BToV)是套式病毒目(Nidovirales)托巴套氏病毒科(Tobaniviridae)凸隆病毒属 (Torovirus)的成员[1],是经证实的牛腹泻病原,主要引起犊牛腹泻甚至死亡,同时也可引起成年牛腹泻[2-3]。该病毒在牛呼吸道中也可检出,具有双重组织嗜性,是潜在的呼吸道致病因子[4-5]。自1982年在美国首次报道以来,目前世界上已有17个国家报道BToV的存在或流行,其已经具有广泛的地域分布[4-14]。
BToV为有囊膜单股正链RNA病毒,基因组由5′UTR和ORF1a、ORF1b、S、M、HE、N 6个ORF区以及3′UTR组成[15]。其中,HE基因编码的血凝素酯酶蛋白(HE蛋白)具有凝集素结构域 R(136—282 aa)、酯酶结构域 E(21—135 aa、283—338 aa)、膜近端结构域 MP(15—20 aa、339—384 aa)3个 结构域[16-17]。在病毒感染的初期,HE蛋白帮助病毒实现与唾液酸的可逆性结合,从而逃避宿主的免疫作用[16,18-19]。这种具有受体破坏酶活性的结构蛋白还在A群β冠状病毒、C型流感病毒、传染性鲑鱼贫血病毒这3类病毒中发现[16,19-21]。在A群β冠状病毒中,血凝素酯酶蛋白不仅作为病毒感染过程中的第二受体,还参与宿主特异性和组织嗜性的改变,并且还能诱导机体产生中和抗体[22-24]。在C型流感病毒中,血凝素酯酶蛋白的变异可能会导致病毒发生抗原漂移[25]。而在传染性鲑鱼贫血病毒中,血凝素酯酶蛋白则决定着病毒的致病性[21]。可见血凝素酯酶蛋白与病毒的毒力改变、组织嗜性改变、和跨种间传播密切相关,在病毒的感染与进化中起着重要作用。
根据HE基因序列特征可将BToV划分为3个基因型(基因Ⅰ~Ⅲ型)[26],当前世界各国主要流行的是Ⅱ型和Ⅲ型[3,8,27]。本实验室最近证明了BToV在国内牛群中的存在,检测到的毒株均为Ⅱ型毒株[8]。目前,BToV在国内牛群中的流行病学资料仍然很少,本研究旨在进一步分析我国BToVHE基因分子特征,为牛腹泻疾病的防控和BToV的遗传进化研究提供参考。
1 材料与方法
1.1 临床样本
94份牛腹泻样本采集于2018年11月—2020年9月,其中,20份犊奶牛(3月龄以内)腹泻样本采自辽宁2个养殖场,58份犊奶牛(3月龄以内)腹泻样本采自河南2个养殖场,16份肉牛(6月龄)腹泻样本采自四川1个养殖场,样本冻存于-80 ℃。
1.2 主要试剂及仪器
TrizolTM、pMD19-T克隆载体、反转录试剂盒、DH5α感受态细胞等购于宝生物工程(大连)有限公司;PCR预混酶Quick Taq HS Dye Mix购于东洋纺(上海)生物科技有限公司;凝胶成像系统Doc2000(Bio-Rad公司,美国);基因扩增仪(ThermoFisher Scientific公司,美国)。
1.3 RNA提取与cDNA合成
取粪便0.2 g与PBS缓冲液(1∶5)混匀后以8 000 r·min-1离心8 min,取上清液400 μL用Trizol 法提取RNA,然后反转录合成cDNA冻存于-20 ℃ 待用。
1.4 临床样本中BToV的检测
采用本实验室建立的RT-PCR方法[8]进行BToV检测。PCR阳性产物送生物工程(上海)股份有限公司测序以验证检测准确性。
1.5 HE基因扩增
根据GenBank中已有的3条完整基因Ⅰ型HE基因序列,15条完整Ⅱ型HE基因序列,3条完整Ⅲ型HE基因序列(参考序列GenBank登录号已在遗传进化树中标出),采用Primer Premier 5.0设计6对引物(表1),用于扩增阳性样本中不同基因型的完整HE基因。PCR扩增条件:94 ℃ 2 min,94 ℃ 30 s,48 ℃ 30 s,68 ℃ 1 min,35个循环。反应体系:QuickTaqHS Dye Mix 12.5 μL,上、下游引物各1 μL,cDNA 2 μL,ddH2O补足25 μL。阳性PCR产物使用胶回收试剂盒回收后连接pMD19-T载体,转入DH5α感受态细胞,增菌后送生工生物工程(上海)有限公司测序。对获得的序列采用SeqMan7.0 (version 7.0;DNASTAR,USA)拼接。
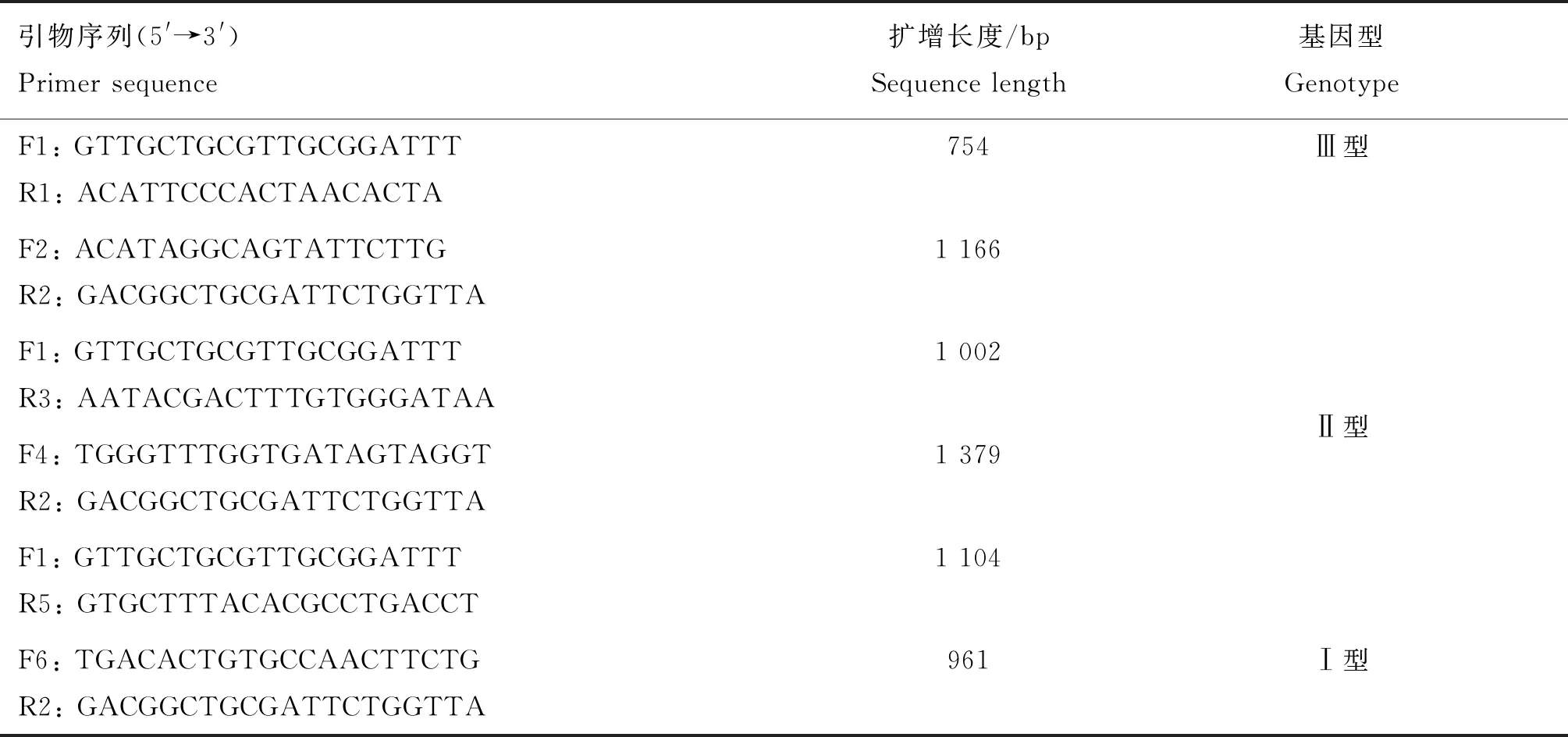
表1 HE基因扩增引物信息Table 1 Primer information of HE gene
1.6 遗传进化和重组及结构分析
使用MEGA 7.0.26进行多序列比对,并采用最大似然法建立氨基酸系统发育树;使用MegAlign (DNASTAR Inc.,WI,USA)计算核苷酸和氨基酸相似性;使用SimPlot 3.5.1和RDP 4.97中的RDP、GeneConv、Chimaera、MaxChi、BootScan、SiScan、3Seq 7种方法进行重组分析;参照牛凸隆病毒血凝素酯酶(SMTL ID:3i26.1)的晶体结构,使用SWISS-MODEL (https://swissmodel.expasy.org/interactive)和UCSF Chimera 1.14构建本研究中HE蛋白的三维模型[16]。
2 结 果
2.1 腹泻样本中BToV检测
从94份牛腹泻样本检出10份BToV阳性,平均阳性率为10.64%,阳性样本分别来自辽宁和河南以及四川的4个牛场;辽宁、河南、四川的阳性率:10%(2/20)、1.72%(1/58)、43.75%(7/16)。
2.2 HE基因扩增和序列分析
从10份阳性样本中成功扩增得到15条完整HE基因,包括9条Ⅱ型(GenBank登录号:MW281062~MW281068、MW114526、MW114527)和6条Ⅲ型(GenBank登录号:MW281069~MW281073、MW114525),其中,四川的5份阳性样本存在基因Ⅱ型和Ⅲ型毒株的混合感染,河南的1份样本单独感染基因Ⅲ型毒株,辽宁和四川的各2份样本单独感染Ⅱ型毒株。
本研究获得的6条完整的基因Ⅲ型HE基因长度均为1 257 bp,编码418个氨基酸,在238位点(相比Ⅱ毒株)缺失1个氨基酸。它们之间的核苷酸相似性为98.6%~100.0%,氨基酸相似性为98.1%~100.0%,与GenBank中已有的21个完整HE基因的核苷酸相似性为71.8%~99.3%,氨基酸相似性为71.2%~99.0%。进一步分析表明,与GenBank中已有的3条国外Ⅲ型毒株HE基因相比,本研究获得的6条Ⅲ型毒株的HE基因具有共同的4个氨基酸位点突变:V78A、E143A、S238T、Y418H,其中,V78A位于酯酶结构域,E143A、S238T则位于凝集素结构域。基于完整HE基因氨基酸序列的系统发育分析表明,本研究获得的6条完整的基因Ⅲ型HE基因则在进化树上聚为独立的1个小支,与土耳其毒株MG957145.1亲缘关系最近(图1)。
本研究获得的9条Ⅱ型HE基因长度均为1 260 bp,编码419个氨基酸,它们之间的核苷酸相似性为96.0%~100.0%,氨基酸相似性为96%~100%,与GenBank中其余的21个完整HE基因的核苷酸相似性为77.1%~99.0%,氨基酸相似性为77.1%~99.0%。与GenBank中已有的15条Ⅱ型HE基因相比,本试验Ⅱ型毒株MW281064、MW114526、MW114527和GenBank中Ⅱ型毒株MN073058、MN073059、AJ575379.1 在凝集素结构域中存在1个 独特的氨基酸突变(T150A)。而本试验Ⅱ型毒株MW281062、MW281063、MW281065~MW281068和土耳其Ⅱ型毒株MG957146.1则具有2个共同的氨基酸突变(I52V、T177N),分别位于凝集素结构域和酯酶结构域。基于完整HE基因氨基酸序列的系统发育分析表明,本研究中获得的9条Ⅱ型HE基因由于上述的氨基酸突变导致它们在遗传进化树上分别聚为2个不同的分支(图1)。
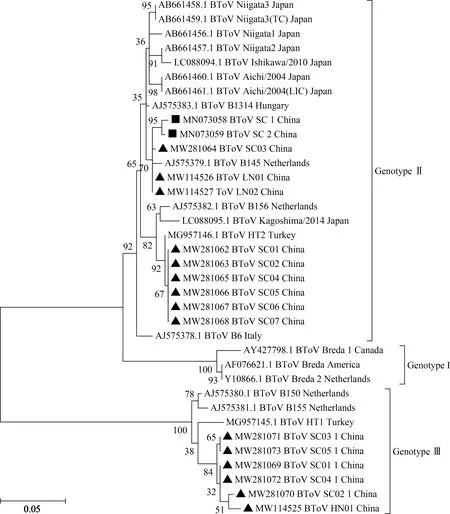
▲.本研究中BToV毒株;■.中国BToV毒株▲.BToV strains in this study;■.BToV strains in China图1 BToV完整HE基因氨基酸序列进化树Fig.1 Phylogenetic tree based on the complete amino acid sequence of HE genes
选取GenBank中39条完整HE基因(包括GenBank中已有的21条BToVHE基因、本研究获得的15条BToVHE基因、3条猪凸隆病毒(PToV)HE基因:AJ575363.1~AJ575365.1)进行重组分析,结果表明,本研究获得的9条Ⅱ型HE基因存在重组事件,预测的重组位点和亲本毒株与Smits等[26]的研究一致。同时6条基因Ⅲ型HE基因也鉴定出重组事件(RDP、GeneConv、Chimaera、MaxChi、BootScan、SiScan、3Seq7种方法支持,得分为0.493),重组区域位于105—1 083 bp,预测的主要亲本毒株是Ⅰ型原型毒株AF076621.1,次要亲本毒株是Ⅱ型毒株AB661461.1(图2)。SimPlot 3.5.1也支持该重组事件;进一步分析显示3个国外Ⅲ型毒株AJ575380.1、AJ575381.1、MG957145.1也具有相同的重组事件。

图2 BToV-MW114525 HE基因重组分析Fig.2 Recombinant analysis the HE gene of BToV-MW114525
根据BToV HE蛋白晶体结构(SMTL ID:3i26.1)构建的三维模型表明,本研究获得的6个Ⅲ型HE蛋白模型在238—240位点处的无规卷曲与其余国外Ⅲ型毒株有差别(图3C)。由于所有Ⅲ型毒株在238位点(相比Ⅱ型毒株)存在1个氨基酸缺失,Ⅲ型毒株在238—240位点的无规卷曲均短于Ⅱ型毒株(图3A、B)。

A.本研究中基因Ⅱ型毒株SC06;B.国外基因Ⅲ型毒株HT1(GenBank:MG957145.1);C.本研究中基因Ⅲ型毒株HN01;蓝色.凝集素结构域(R);绿色.酯酶结构域(E);红色.膜近端结构域(MP);虚线框内表示有改变的空间结构Predicted crystal structures of the BToV HE proteins from three strains:A.Genotype Ⅱ strain SC06 in this study;B.Genotype Ⅲ strain HT1 (GenBank accession No.MG957145.1);C.Genotype Ⅲ strain HN01 in this study.The domains are color-coded:lectin domain (R,blue);esterase domain (E,green);membrane-proximal domain (MP,red).The boxed portions of the structures indicate the different conformations of the same R domain of BToV strains图3 基于BToV HE蛋白晶体结构(SMTL ID:3i26.1)构建的三维模型Fig.3 The 3D models constructed based on the crystal structure of BToV hemagglutinin-esterase (SMTL ID:3i26.1)
3 讨 论
BToV是导致牛腹泻的重要病原,主要引起犊牛腹泻,同时也可引起成年牛腹泻,造成严重的经济损失[2,3,6,18,28]。BToV在我国属于新发牛腹泻病原,本实验室前期报道了BToV在国内的存在和流行,均为基因Ⅱ型毒株[8],继获得牦牛源BToV基因Ⅲ型基因组[29]后,本研究首次报道了基因Ⅲ型毒株在我国牛群中的存在,为了解国内BToV的遗传多样性提供了有用的信息。本试验鉴定了Ⅲ型和Ⅱ型毒株的混合感染,这是首次观察到BToV不同基因型混合感染现象,有助于进一步了解BToV的感染和流行特征。系统发育分析表明,本研究获得的9条Ⅱ型毒株分为2个不同的分支(图1),不同分支上的毒株各自产生了独特的氨基酸突变,显示出国内基因Ⅱ型毒株具有遗传多样性。而6条Ⅲ型毒株则独立聚为1支,显示出Ⅲ型毒株在我国具有独特的遗传进化趋势(图1)。由于样本数量有限,不同基因型毒株在我国的流行情况还需进一步调查。
BToV HE蛋白具有凝集素结构域(136—282aa)、酯酶结构域(21—135 aa、283—338 aa)和膜近端结构域(15—20 aa、339—384 aa)3个结构域[16-17],其中,凝集素结构域介导病毒与宿主细胞的唾液酸结合,HE蛋白的238—240位点位于凝集素结构域空间结构的最外侧,可能在病毒结合受体的过程中起着重要作用[16]。而酯酶结构域则发挥受体破坏酶活性,使病毒可逆性黏附唾液酸,帮助病毒逃避宿主的体液免疫[16-17]。与GneBank中仅有的国外3条Ⅲ型HE基因相比,本试验获得的6条Ⅲ型HE基因具有共同的2个氨基酸位点突变(E143A、S238T)位于HE蛋白的凝集素结构域[16-17],特别238位氨基酸位点由丝氨酸突变为苏氨酸引起238—240位点处的无规卷曲也发生了改变,这可能会影响病毒与宿主唾液酸的结合[16-17]。此外,获得的6条Ⅲ型HE基因在酯酶结构域还有1个相同的氨基酸突变(V78A),是否会影响病毒与唾液酸的可逆性黏附有待于进一步研究[16-17]。
相比Ⅰ型和Ⅱ型毒株,所有Ⅲ型毒株(包括本研究毒株)在凝集素结构域中存在2个独特的氨基酸突变(G210D、L170V),而该位点是形成结合受体的“疏水袋”关键位点[16-17],这可能就会改变整个基因Ⅲ型BToV毒株与唾液酸受体的结合能力,进而影响病毒与宿主细胞的结合[16-17]。在酯酶结构域中,所有Ⅲ型毒株(包括本研究毒株)的2个位点产生了突变(R103H、Y294R),由于该位点与HE蛋白结合唾液酸类型有关[16-17],因此,这可能会改变基因Ⅲ型BToV毒株的唾液酸的结合类型,从而影响基因Ⅲ型BToV毒株的组织噬性。而相比Ⅰ型和Ⅲ型,所有Ⅱ型毒株(包括本研究毒株)则发生了1个氨基酸突变(L168F),该位点也是形成结合受体的“疏水袋”关键位点[16-17],可能会影响Ⅱ型BToV毒株与宿主细胞受体的结合能力。最早报道BToV的毒株均为Ⅰ型毒株[15,30],但目前各国流行的却是Ⅱ型和Ⅲ型毒株[3,8,27],这种流行情况的改变可能与这些氨基酸突变有关,值得进一步调查。
Smits等[26]于2003年的研究显示,基因Ⅱ型BToV毒株是由原型(Ⅰ型)毒株与PToV在HE基因的3′端发生重组而来,而Ⅲ型BToV毒株可能是Ⅱ型BToV毒株与一种未知的ToV在HE基因的中间区域发生重组而来。本研究预测的Ⅱ型毒株重组事件与先前研究结果一致[26],但Ⅲ型毒株的重组事件所预测的亲本毒株与先前研究结果不同[26]。本研究结果显示,Ⅲ型毒株是由Ⅰ型毒株和Ⅱ型毒株重组而来,预测出的亲本毒株为Ⅰ型毒株AF076621.1和Ⅱ型毒株AB661461.1,重组区域位于105—1 083 bp,并且GneBank中其余Ⅲ型毒株均具有相同重组事件。造成该重组分析结果不同的原因是本研究在做重组分析时加入了GneBank中近年新登录的BToVHE基因序列。这种通过HE基因同源重组产生新基因型毒株可能是病毒在进化过程中逃避宿主免疫的一种策略[18-19,31],这对进一步了解BToV的遗传进化有重要意义。
4 结 论
本研究证实了基因Ⅲ型BToV在我国牛群中的存在,首次鉴定了BToV不同基因型毒株的混合感染,为国内牛腹泻防控提供了参考。本研究获得的6条Ⅲ型HE基因存在4个独特的氨基酸突变,其中2个突变位点位于凝集素结构域,1个突变位点位于酯酶结构域,可能会影响病毒与受体的结合。重组分析显示Ⅲ型BToV毒株可能是Ⅱ型BToV毒株与Ⅰ型BToV毒株重组而来,对进一步了解BToV的遗传进化有重要意义。
