羟基积雪草苷对大鼠创伤性颅脑损伤后神经功能障碍的改善作用及机制研究
2021-07-23贾颜锋
贾颜锋,陈 伟
1.西安交通大学第一附属医院神经外科,西安 710061;2.西安交通大学附属西安市人民医院神经外科,西安 710004
在创伤性颅脑损伤(traumatic brain injury,TBI)后继发的复杂病理生理进程中,受损脑组织产生的兴奋性氨基酸过量聚集,可通过作用于神经元细胞膜N-甲基-D-天门冬氨酸 (N-methyl-D-aspartic acid,NMDA) 受体,引起兴奋性神经毒损伤和细胞凋亡,是造成TBI后神经功能障碍的重要因素[1-2]。研究表明,细胞表面的NMDA受体2B亚基 (NMDA receptor 2B,NR2B) 磷酸化水平与表达量越高,其对兴奋性氨基酸的毒损伤易感性越强[3-4]。因此,抑制TBI后NR2B与磷酸化NR2B (Phosphorylated NR2B,pNR2B) 表达水平,可能是阻止NMDA受体介导的兴奋性神经毒损伤以及连锁效应的有效途径。
羟基积雪草苷(madecassoside,MC)是提取自伞形科植物积雪草的三萜类化合物,是积雪草的主要药理活性成分,具有抗氧化应激、抗炎、促进创面愈合等多种功效[5-6]。新近研究发现,MC对中枢神经系统退行性病变和缺血性疾病具有神经保护作用[7]。然而,MC对颅脑损伤的实验研究未见报道。本文通过建立大鼠TBI模型,观察MC对颅脑损伤后神经功能障碍的改善作用,并从兴奋性毒损伤效应引起神经元凋亡的角度初步探讨MC的可能作用机制,为其应用于TBI的脑保护治疗提供新的研究依据。
材料与方法
1 主要设备与试剂
控制性皮层损伤打击装置(美国Hatteras 公司);免疫印迹电泳装置(美国Bio-Rad公司);NR2B抗体和pNR2B抗体(武汉博士德生物工程有限公司);B细胞淋巴瘤-2(B cell lymphoma,Bcl-2)基因抗体、半胱天冬酶-3(Caspase-3)抗体和Bcl-2相关X蛋白(Bcl-2 associated X,Bax) 抗体(美国CST公司);β-actin抗体(美国Thermo Fisher公司);Western-blotting检测试剂盒和BCA蛋白定量试剂盒(北京碧云天生物科技有限公司);羟基积雪草苷 (南京道斯夫生物科技有限公司)。
2 实验分组与给药方法
SPF级雄性SD大鼠64只(200~220g/只),由西安交通大学医学部实验动物中心提供。采用随机数字表法将大鼠分为四组:假创伤组、创伤组、创伤后低剂量MC治疗组(25mg/kg)、创伤后高剂量MC治疗组(75mg/kg)。MC溶解于0.9%氯化钠注射液中,低剂量MC组与高剂量MC组大鼠在伤后1h时尾静脉注射给药干预,此后每日在相同时点尾静脉给药1次,连续28d。本研究大鼠饲养和实验过程均严格遵守实验动物伦理规定,获西安交通大学医学部动物实验伦理委员会批准(XJTU-M202001295a)。
3 建立大鼠TBI模型
以2%戊巴比妥钠溶液腹腔注射麻醉大鼠(60mg/kg),沿中线纵行切开头顶皮肤,剥离骨膜,于人字缝尖与前囟的中点右偏4mm处,牙科钻开一直径为3mm的颅骨骨窗,显露硬脑膜。连接控制性皮层损伤装置,将大鼠固定于打击台上,调整打击棒位置,使其尖端接触至硬脑膜表面,实施皮层打击致伤,打击速度为4.0m/s、致伤深度2.0mm、接触时间为120ms,完成打击后予以缝合头皮。假损伤组大鼠仅纵形切开头皮后缝合,不予以实施打击。
4 蛋白免疫印迹检测(Western-blotting)
大鼠伤后28d处死,采用Western-blotting检测各组大鼠皮层损伤区NR2B、 pNR2B、Bcl-2、Caspase-3和Bax的表达水平。以2%戊巴比妥钠溶液腹腔注射麻醉大鼠(60mg/kg),快速断头取脑,冰上分离获得皮层损伤区脑组织。冰浴条件下裂解、匀浆、提取总蛋白。采用蛋白质定量法(BCA法)蛋白定量后,上样进行SDS聚丙烯酰胺凝胶电泳、转膜。5%脱脂奶粉溶液封闭1h后,TBST洗涤3次,每次8min。分别加入NR2B一抗(1∶1000)、pNR2B一抗(1∶800)、Bcl-2一抗(1∶2000)、Caspase-3一抗(1∶1000)、Bax一抗(1∶2000)和β-actin一抗(1∶1500),4℃条件下反应过夜,TBST洗涤3次,每次8min。再加入IgG二抗(1∶2000),室温条件下反应1h,TBST洗涤3次,每次8min。加入显影液进行发光,凝胶成像系统分析,测定各目标条带与内参β-actin条带的灰度,得出相对灰度比值。
5 大鼠神经功能评分
伤后3、7、14、21和28d,采用改良神经功能缺损严重程度评分量表(modified neurological severity score,mNSS) 分别对各组大鼠神经功能进行评估。根据该量表评分标准与步骤,依次在不同时点对各组大鼠进行运动实验、感觉实验、平衡木实验和反射实验[8]。综合各项实验得分,分数越高代表神经功能缺损越严重,1~6分为轻度功能缺损,7~12分为中度功能缺损,13~18分为重度功能缺损。
6 统计学分析
结 果
1 大鼠神经功能评分比较
伤后3、7、14、21和28d,创伤组、低剂量MC组与高剂量MC组大鼠在各时点的mNSS评分均高于假创伤组(P<0.05)。伤后3d,创伤组、低剂量MC组与高剂量MC组的评分差异无统计学意义(P>0.05)。伤后7、14、21和28d,低剂量MC组与高剂量MC组的评分均显著低于创伤组(P<0.05),并且高剂量MC组评分显著低于低剂量MC组,组间差异有统计学意义(P<0.05)。此外,在伤后7、14、21和28d,低剂量MC组与高剂量MC组的评分随着时间延长逐渐降低,两组内各时点间的差异也有统计学意义(P<0.05)。见表1。
表1 各组大鼠伤后各时点的mNSS评分比较分)
2 大鼠皮层损伤区NR2B与pNR2B表达水平
伤后28d,创伤组大鼠皮层损伤区的NR2B与pNR2B表达量较假创伤组显著增加。与创伤组相比,低剂量MC组、高剂量MC组两个给药组的NR2B与pNR2B表达水平明显降低。并且高剂量MC组的NR2B与pNR2B表达水平均显著低于低剂量MC组,组间差异有统计学意义(P<0.05)。见图1、表2。
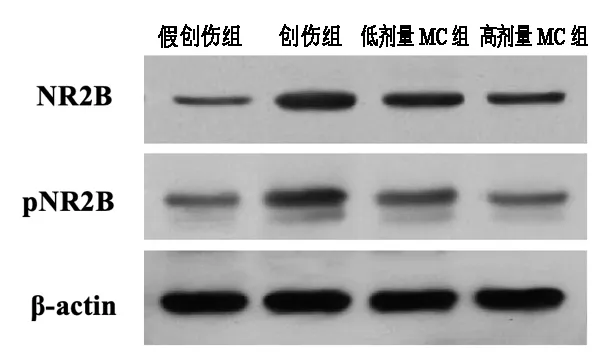
图1 各组大鼠皮层区NR2B与pNR2B表达检测结果

表2 各组大鼠NR2B与pNR2B表达量比较
3 大鼠皮层损伤区Caspase-3、Bax与Bcl-2表达水平
与假创伤组相比,创伤组大鼠皮层损伤区域的Caspase-3、Bax与Bcl-2的表达量均增加。与创伤组相比,低剂量MC组与高剂量MC组的Caspase-3与Bax表达水平减少,Bcl-2表达水平明显增加。两个给药组相比,高剂量MC组Caspase-3与Bax的表达水平显著低于低剂量MC组,Bcl-2的表达水平高于低剂量MC组,组间差异存在统计学意义(P<0.05)。见图2与表3。
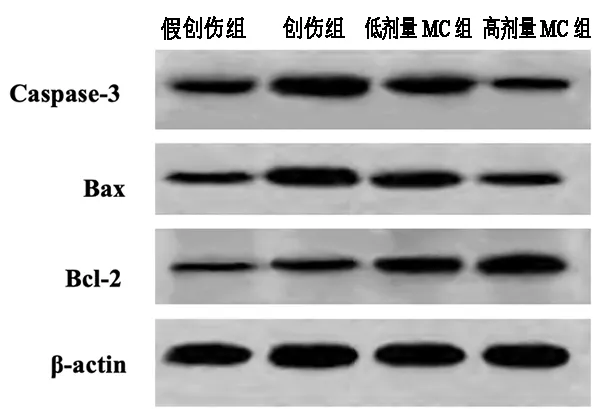
图2 各组大鼠皮层区Caspase-3、Bax与Bcl-2表达检测结果
表3 各组Caspase-3、Bax与Bcl-2表达量比较
讨 论
随着现代社会和交通工具的发展,TBI作为常见的急性创伤,发生率逐渐升高。TBI以高致残率和致死率居各类创伤之首,已成为严重威胁公共健康的卫生问题。虽然救治技术的进步使TBI病死率显著下降,但对幸存患者的神经功能障碍救治难以令人满意,尤其是对TBI后多因素参与的继发性脑损伤缺乏有效干预措施[9]。因此,深入研究TBI后继发性脑损伤的致伤因素与机制,探讨合理可行的治疗策略,对改善神经功能缺损和促进患者康复具有重要意义。本研究利用控制性皮层损伤法制备TBI模型,初步发现MC对TBI具有神经保护作用。
MC作为传统中药积雪草的主要药理成分,既往研究证实其对中枢神经系统多种退行性疾病具有神经保护作用[5]。MC可显著提高侧索硬化小鼠的脊髓前角神经元数量,减少神经元变性,有效改善侧索硬化小鼠的神经功能缺陷[7]。MC通过抑制1甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)对多巴胺能神经元的毒性损伤作用,减少神经元的凋亡,增加纹状体多巴胺的含量,显著改善帕金森大鼠的运动功能障碍[10-11]。MC还可抑制脑缺血再灌注损伤时小胶质细胞向促炎表型极化,减轻小胶质细胞介导的炎症反应,减少促炎因子的释放和自由基的产生[12-13]。大鼠脊髓损伤后,MC干预可显著提高脊髓组织的超氧化物歧化酶活性,减轻损伤脊髓的水肿程度和神经元凋亡数量[6]。
在TBI后继发性脑损伤的病理生理变化中,损伤区域脑组织兴奋性氨基酸大量聚集,结合至位于神经元细胞膜的N-甲基-D-天冬氨酸(NMDA)受体,而引起兴奋性神经毒损伤效应,导致神经元细胞凋亡、坏死,进而加重继发性脑损害[2-4]。构成NMDA受体的多个亚单位中,主要调节亚单位NR2包括NR2A、NR2B、NR2C和NR2D四个亚基。其中,NR2B亚基作为调控NMDA受体功能与活性的主要分子,其磷酸化是NMDA受体介导兴奋性神经毒损伤过程中的关键环节,直接决定了神经元细胞对兴奋性氨基酸毒损伤效应的易感性[14-15]。研究表明,抑制NR2B的表达及其磷酸化水平,可有效减轻兴奋性氨基酸堆积引起的细胞外钙离子内流超载,以及最终导致的兴奋性毒损伤效应[4,16]。
本研究利用控制性皮层损伤法建立TBI大鼠模型,首先通过mNSS神经功能缺损评分,证实了TBI引起的运动、感觉、反射等神经功能障碍。给予MC治疗发现受伤大鼠的神经功能障碍明显改善,而且高剂量MC治疗组的效果显著优于低剂量MC治疗组。皮层损伤区NR2B与pNR2B的检测结果发现,TBI后NR2B表达水平及其磷酸化水平显著增加,表明伤后皮层区NR2B被活化,引起兴奋性神经毒性损伤,可能是导致TBI后神经功能障碍和加重继发性脑损伤的重要因素。给予MC干预治疗,可显著抑制NR2B的表达及其磷酸化,改善大鼠的神经功能障碍。同样,笔者发现,高剂量MC对NR2B介导的兴奋性神经毒性损伤抑制效果显著优于低剂量。
神经元凋亡是受兴奋性毒损伤后的主要死亡原因之一,抑制TBI后神经元细胞凋亡发生、发展,可有效减轻继发性神经损害和促进功能恢复[17-18]。在目前已知的众多调节细胞凋亡分子中,Bcl家族的促进凋亡基因Bax和抑制凋亡基因Bcl-2是一对极为重要的调控分子。Bax与Bcl-2在凋亡进程中的对立功能,使二者的表达水平直接决定了细胞凋亡阈值[19]。在凋亡信号刺激下,Bax构象发生改变,并转位至线粒体破坏膜通透性,引起大量促凋亡因子释放,激活Caspase-3,进而启动细胞凋亡和加速凋亡进程[20]。Bcl-2作为抑制细胞凋亡基因,通过与Bax形成异源二聚体,可抑制其转位以及活化Caspase-3的级联反应,有效阻止细胞凋亡程序启动[21]。Bcl-2还可通过调节内质网中钙离子流向,稳定胞内钙离子浓度,组织钙离子超载进一步加重,阻断兴奋性毒损伤诱发的细胞凋亡进程[19-21]。
如何减轻兴奋性毒损伤引起的神经元凋亡,是近年来TBI的脑保护策略研究热点。本研究在明确TBI后皮层损伤区NR2B介导的兴奋性毒损伤基础上,进一步对该区域凋亡相关分子进行检测,结果提示伤后促凋亡分子Caspase-3和Bax表达明显增加,抗凋亡分子Bcl-2表达显著下降,表明NR2B活化引起的兴奋性毒效应可诱导神经元细胞凋亡。伤后予以大剂量和小剂量MC干预治疗,均能够有效抑制NR2B活化和神经毒性反应,下调促凋亡分子表达和提高抗凋亡分子的表达,逆转TBI后兴奋性毒损伤引起的神经元凋亡,并且高剂量组的效果优于低剂量组。据此,笔者推测,MC对颅脑损伤的神经保护作用,可能与减轻NR2B介导的兴奋性神经毒损伤及其引起的神经元凋亡有关。
综上所述,本研究通过建立大鼠TBI模型和给予MC干预治疗,发现TBI后MC可有效抑制兴奋性神经毒损伤的重要诱导分子NR2B活化,减轻皮层区神经元凋亡,改善神经功能障碍,初步证实了MC对TBI具有神经保护作用,为其向临床转化应用提供了更加充分的实验依据。但TBI后MC的最佳治疗剂量、给药时间窗和具体作用途径等仍有待进一步研究。
