基于高效液相色谱的BPTAP 纯度分析方法
2021-07-22陈建波赵海霞
丁 欢,陈建波,赵海霞,刘 渝
(1. 中国工程物理研究院化工材料研究所,四川 绵阳 621900;2. 中北大学环境与安全工程学院,山西 太原 030051)
1 引言
耐热炸药作为一类热安定性能优异的含能材料,不仅具有低的感度和高的能量输出,还具有高的熔点(200 ℃以上)和低的蒸汽压,在长期的高温加载后仍能可靠起爆[1-2]。根据耐热性能的不同,耐热炸药可分为高温耐热炸药(耐热温度低于220 ℃)和超高温耐热炸药(耐热温度高于250 ℃),其中超高温耐热炸药更能适应严苛的作战环境,是含能材料领域未来的发展趋势之一[3-4]。四硝基苯并-1,3a,6,6a-四氮杂戊搭烯并吡啶(BPTAP)的分子结构为大π键的共轭稠环体系,使其具有高的热分解温度(375 ℃),且熔点大于300 ℃[5-6]。此外,BPTAP 还具有低的感度(摩擦感度为120 N,撞击感度为26 N)和高的能量(爆速为7.43 km·s-1,爆压为29.4 GPa)[7]。鉴于BPTAP 优异的性能,被认为是新一代的超高温耐热炸药,近年来受到含能材料研究人员的广泛关注。
纯度是炸药品质评价的重要指标之一,常用的纯度测试方法主要包括凝固点下降法[8]、差热分析法(DSC)[9]、滴定法[10]、核磁共振氢谱法(1H NMR)[11]、色谱法[12]/高效液相色谱(HPLC)等,其中HPLC 具有样品用量少、灵敏度高、重现性好、自动化程度高等优势,基于HPLC 的纯度分析方法是有机化合物纯度分析的主要研究手段,在含能材料领域有着广泛的应用。刘秀华等[13]针对8 种硝基化合物建立了基于HPLC 的含量分析方法,不但可同时检测多种炸药组分,还具有高的检测灵敏度(检测限≤0.8 ng)。赵允颖等[14]基于HPLC 方法实现了六硝基茋(HNS)与其杂质六硝基联苯(HNBB)的有效分离与定量分析,对HNS 和HNBB 的 检 测 范 围 分 别 为5~100 mg·L-1和2~50 mg·L-1,满 足 了HNS 及 其 相 关 物 质 的 质 量 控制需求。刘园园等[15]针对3,4-二硝基吡唑及其杂质3-硝基吡唑和1,3-二硝基吡唑,建立了基于HPLC 的纯度分析方法,三种化合物的检测限分别为1.19,0.60 mg·L-1和1.04 mg·L-1。可 见,HPLC的众多优点使其成为含能材料定性定量分析的重要手段之一,在含能材料的研制生产过程中发挥了重要作用。
BPTAP 作为近年来高度关注的一类新型耐热炸药,人们围绕BPTAP 开展了一些合成与性能研究[16-17],正努力推进其工程化应用,然而BPTAP 目前仍缺乏相应的纯度分析方法,不利于其研制过程的纯度监测与质量控制。为此,本研究开展了基于反相高效液相色谱(RP-HPLC)的BPTAP 纯度分析方法研究,通过建立相应的色谱分析方法,为BPTAP 工程化应用的纯度分析与质量控制提供技术支撑。
2 实验部分
2.1 实验仪器
Shimadzu LC-20AT 高效液相色谱仪,配置有2 个LC-20AT 泵,SIL-20A 自动进样器,SPD-20A 紫外检测器,CTO-20A 柱温箱。Agilent 1260 液相色谱仪,配置有DAD 检测器。超纯水出自Millipore-Q system(美国Millipore),电阻率为18.2 MΩ。炸药样品均在XPE26 微量天平(瑞士Mettler Toledo)上称重,精度为0.001 mg。UA10MFD 超声波清洗器(德国WIGGENS),Vortex Genie 2T 旋涡混合器(美国Scientific Industries),BL-240/2411 防爆冰箱(上海亿思科技实业有限公司)。
2.2 试剂与耗材
BPTAP 炸药由中物院化工材料研究所提供,并进行重结晶(纯度≥98.0%);甲醇(HPLC 级,百灵威化学试剂公司),乙腈(HPLC 级,百灵威化学试剂公司),四氢呋喃(HPLC 级,默克化工技术公司),乙酸铵(HPLC级,百灵威化学试剂公司),三氟乙酸(AR级,麦克林生化公司),N',N'-二甲基甲酰胺(AR 级,麦克林生化公司),二甲基亚砜(AR 级,天津市致远化学试剂有限公司);注射器(美国赛默飞世尔实验器材公司),0.46 μm 的过滤膜(日本AS One)。
2.3 样品前处理
准确称取2.00 mg 的BPTAP 样品,置于100 mL 的容量瓶中,乙腈定容,超声搅拌使其充分溶解,配制成20.00 μg·mL-1的溶液,用于HPLC 分析方法开发研究。称取10.06 mg 的BPTAP 纯度参比样品,溶于50 mL 的DMSO 中,配制成201.20 μg·mL-1的储备液;再分别对上述储备液进行稀释,配制成100.60、50.30、10.06、5.03、1.01 μg·mL-1和0.50 μg·mL-1等不同浓度的标准工作溶液,用于标准工作曲线、检测限等相关研究。为调节HPLC 流动相的pH 值,在水相中加入不同类型的添加剂,分别配制成含有0.05%(V/V)三氟乙酸、0.5%(V/V)四氢呋喃和1.5、1.0、0.5 mg·mL-1乙酸铵的流动相。所有样品在HPLC 分析前,均经过0.45 μm 的过滤膜过滤,避免不溶物堵塞色谱柱。
2.4 HPLC 方法的优化
HPLC 分析方法开发主要在Shimadzu LC-20AT高效液相色谱仪上进行,检测波长的优化在Agilent 1260 液相色谱仪上进行。色谱柱是影响HPLC 分离效果好坏的关键因素之一,其中Plus C18 色谱柱适用于大多数有机化合物的分离,应用范围最为广泛。另外,Extend C18 色谱柱适用于碱性条件下分离,Phenyl-Hexyl 色谱柱适于分离芳香族化合物,SB-Phenyl 色谱柱适合在酸性条件下分离芳香族化合物。优化后的HPLC 方法如下:色谱柱为ZORBAX Eclipse Plus C18色谱柱(4.6×150 mm,5.0 μm),流动相为超纯水(含0.1 mg·mL-1的乙酸铵)-乙腈混合液,梯度洗脱模式,乙腈含量在0~10 min 之间由35%线性增至90%并继续保持5 min,从15.01 min 起,乙腈含量回到35%并保 持6 min;流 速 为1.0 mL·min-1,进 样 体 积 为10.0 μL,柱温为30 ℃,检测波长为230 nm。HPLC 分析、数据采集和定量分析均在Shimadzu LCsolution 软件上进行。
2.5 HPLC 方法验证
通过考察BPTAP 色谱分析的重复性、稳定性、精密度、耐用性、标准曲线与灵敏度、检测限等参数分析,对建立的HPLC 分析方法的性能进行验证。
通过重复多次进样(n=6),分析BPTAP 的保留时间和峰面积等相关参量的相对标准偏差(RSD),评估HPLC 方法的重复性。
通过HPLC 在48 h 内不同时间间隔的连续分析,检测不同时间段内BPTAP 的保留时间和峰面积的变化,考察该方法的稳定性。
基 于 不 同 浓 度(201.20、100.60、50.30、10.06、5.03、1.01 μg·mL-1和0.50 μg·mL-1)的BPTAP 标 准溶液建立相应的标准工作曲线,以三倍的信噪比来分析方法的检测限(LOD),以十倍的信噪比分析该方法的定量限(LOQ),以评价该方法的灵敏度。
为验证方法的准确性,在未知的样品中添加不同浓度(200、100、50 μg·mL-1)的BPTAP 纯度参比物,每个样品重复测定三次并计算加标回收率。
通过分析BPTAP 在日间(n=18,每天进样6 次)和日内(n=6)峰面积的RSD 以评估色谱方法的日间/日内精密度。
为研究方法的耐用性,对检测波长±2.0 nm、柱温±2.0 ℃等进行细微调整,考察BPTAP 的保留时间(tR)、分离度(R)和半峰宽(full width at half maxima,简称FWHM)等参量的变化。
2.6 BPTAP 重结晶过程的纯度分析
将BPTAP 粗产品加入DMF 中,80 ℃油浴加热,500 r·min-1搅拌至充分溶解,冷却到室温加入反溶剂(甲醇)搅拌至样品析出,静置过滤,使用超纯水洗涤,干燥得到一次重结晶的样品。油浴温度下降到50 ℃,将一次重结晶的样品按照相同的方法进行二次重结晶。
准确称取BPTAP 粗品、一次重结晶样品和二次重结晶样品各1.00 mg,分别置于100 mL 的容量瓶中,乙腈定容,超声搅拌使其充分溶解,经0.45 μm 的过滤膜过滤,并利用建立的HPLC 方法分析BPTAP 重结晶前后的纯度变化。
3 结果与讨论
3.1 色谱分析影响因素考察
BPTAP 的结构如图1 所示,鉴于BPTAP 在高极性溶剂(如乙腈、甲醇)中的溶解性明显优于低极性溶剂(如正己烷、氯仿),优先采用HPLC 的反相模式进行分析方法开发研究。由于BPTAP 合成得到的粗产品中存在多种杂质,采用梯度洗脱的分离效果明显优于等度洗脱。因此,本研究将基于梯度洗脱模式的RP-HPLC 开展对BPTAP 纯度分析方法研究,重点考察色谱柱、流动相、进样量、流速等因素对分离结果的影响。

图1 BPTAP 的分子结构Fig.1 Molecular structure of BPTAP
3.1.1 色谱柱的优化
本研究考察了四种色谱柱对BPTAP 的色谱分析的影响,结果如图2a所示。由图2a可知,Extend C18色谱柱的分离效果较差,杂质峰都集中于BPTAP 色谱峰附近。Plus C18(4.6 mm×150 mm,5 μm)、Phenyl-Hexyl(4.6 mm×150 mm,5 μm)和SB-Phenyl(4.6×250 mm,5 μm)色谱柱的分离效果有所改进,BPTAP 与其杂质均能达到较好的分离。但是,不同的色谱柱在分离时间上差异较大,SB-Phenyl 色谱柱的分离时间最长(12.04 min),Phenyl-Hexyl 色 谱 柱 的 分 离 时 间 为10.46 min,Plus C18 色谱柱的分离时间较短,为8.99 min。另外,不同色谱柱下BPTAP的色谱峰面积也有所不同,SB-Phenyl色谱柱的峰面积(555158 mV·min)最强,Phenyl-Hexyl 色谱柱的峰面积(531337 mV·min)次之,Plus C18 色谱柱的峰面积(514818 mV·min)较强,而Extend C18 色谱柱的峰面积(309258 mV·min)最弱。为了在提高分离效率的同时达到好的分离效果,综合考虑BPTAP 的保留时间、峰高和峰面积参数,最终选择Plus C18(4.6 mm×150 mm,5 μm)色谱柱进行BPTAP 纯度分析。

图2 BPTAP 在不同色谱柱(a)和不同流动相(b)下的色谱图Fig.2 Chromatographic analysis of BPTAP by using different columns(a)and different mobile phase(b)
3.1.2 流动相的优化
流动相作为色谱分离的重要载体,对分析过程有着重要影响。反相液相色谱的流动相通常由有机相和水相混合而成,其中常见的有机相为乙腈、甲醇等。考察以乙腈和甲醇为流动相时,不同有机溶剂对BPTAP色谱分离的影响,结果如图2b 所示。当以甲醇为有机相时,BPTAP 的保留时间(6.83 min)较短,但与其相邻的色谱峰发生重叠,未能充分分离。当流动相中有机溶剂改为乙腈时,BPTAP 的保留时间(9.36 min)有所延长,但分离效果明显改善,对称性较好,且BPTAP 的峰面积(637343 mV·min)和峰高(67304 mV)信号更强。可见,乙腈作为有机相更适合BPTAP 的色谱分离。
3.1.3 乙腈含量的优化
在梯度洗脱模式下,从起始点到终点过程中有机相与水相的变化比例是决定分离效果的关键因素。首先考察了梯度起点乙腈含量分别为30%、35%、40%、45%和50%时,对BPTAP分离的影响,结果如图3所示。乙腈含量越高,BPTAP的保留时间越短(见图3b)。乙腈含量为25%时保留时间为20.78 min,当乙腈含量升高至50%时,BPTAP 的保留时间缩短至14.36 min。从图3a 可以看出,当乙腈含量为35%时,色谱峰1、2和3 的分离效果最好。随着乙腈含量的持续增加,色谱峰1、2 和3 对应的分离度降低,甚至难以分开。可见,以35%乙腈含量作为BPTAP 梯度分离的起点具有更好的分离效果。

图3 梯度起点不同乙腈含量下的BPTAP 色谱分析Fig.3 Chromatographic analysis of BPTAP by using different acetonitrile content at the starting point
在对梯度起点的乙腈含量优化后,进一步考察了梯度终点不同乙腈含量(分别为75%、80%、85%、90%和95%)对BPTAP 分离的效果。如图4 所示,BPTAP 的保留时间随乙腈含量的增加而略微减小,当终点乙腈含量从75%升至95%时,其保留时间下降了1.198 min,但峰高从43602 mV 增加至48412 mV。当梯度终点乙腈含量为90%时,BPTAP 及其杂质的色谱峰的半峰宽均小于0.30,拖尾因子在1.047~1.228 之间,理论塔板数均大于40703,对称性好,柱效高。因此,选择90%作为BPTAP 梯度分析终点的乙腈含量。
3.1.4 流动相中添加剂的影响
当流动相中未加入添加剂时,保留时间为2 min处存在数个难以分离的色谱峰。为改善分析效果,考察了在流动相中加入不同添加剂对BPTAP 分离的影响。如图5a 所示,加入0.05%(V/V)三氟乙酸时,基线漂移严重;加入0.5%(V/V)四氢呋喃时,对色谱分离效果没有明显改善;而加入1.5 mg·mL-1的乙酸铵时,保留时间为2 min 处各峰的分离有一定改善。进一步考察了不同浓度的乙酸铵(浓度分别为0.5、1.0 和1.5 mg·mL-1)对这些峰的分离效果。如图5b 所示,1.0 mg·mL-1的乙酸铵作为添加剂即可达到较好的分离。因此,以1.0 mg·mL-1的乙酸铵作为流动相中的添加剂。

图5 流动相中不同的添加剂对BPTAP 色谱分析的影响Fig.5 Chromatographic analysis of BPTAP by using different additives in mobile phase
3.1.5 检测波长的优化
利用带有二极管阵列检测器的液相色谱考察不同检测波长对BPTAP 分离的影响。如图6所示,当以BPTAP的最大紫外吸收波长460 nm 为检测波长时,BPTAP 具有最大的峰高和峰面积。同时,以200~230 nm 为检测波长时,BPTAP 也有较强的色谱信号。此外,相对于460 nm 的检测波长,200~230 nm 的检测波长下能看到更多的杂质峰,更有利于BPTAP 制备过程的杂质控制。然而,当以200 nm、210 nm 为检测波长时,基线漂移严重,背景噪音较强。而在230 nm 检测波长时,基线更为平滑。因此,选择230 nm 作为BPTAP 色谱分析的最佳检测波长。
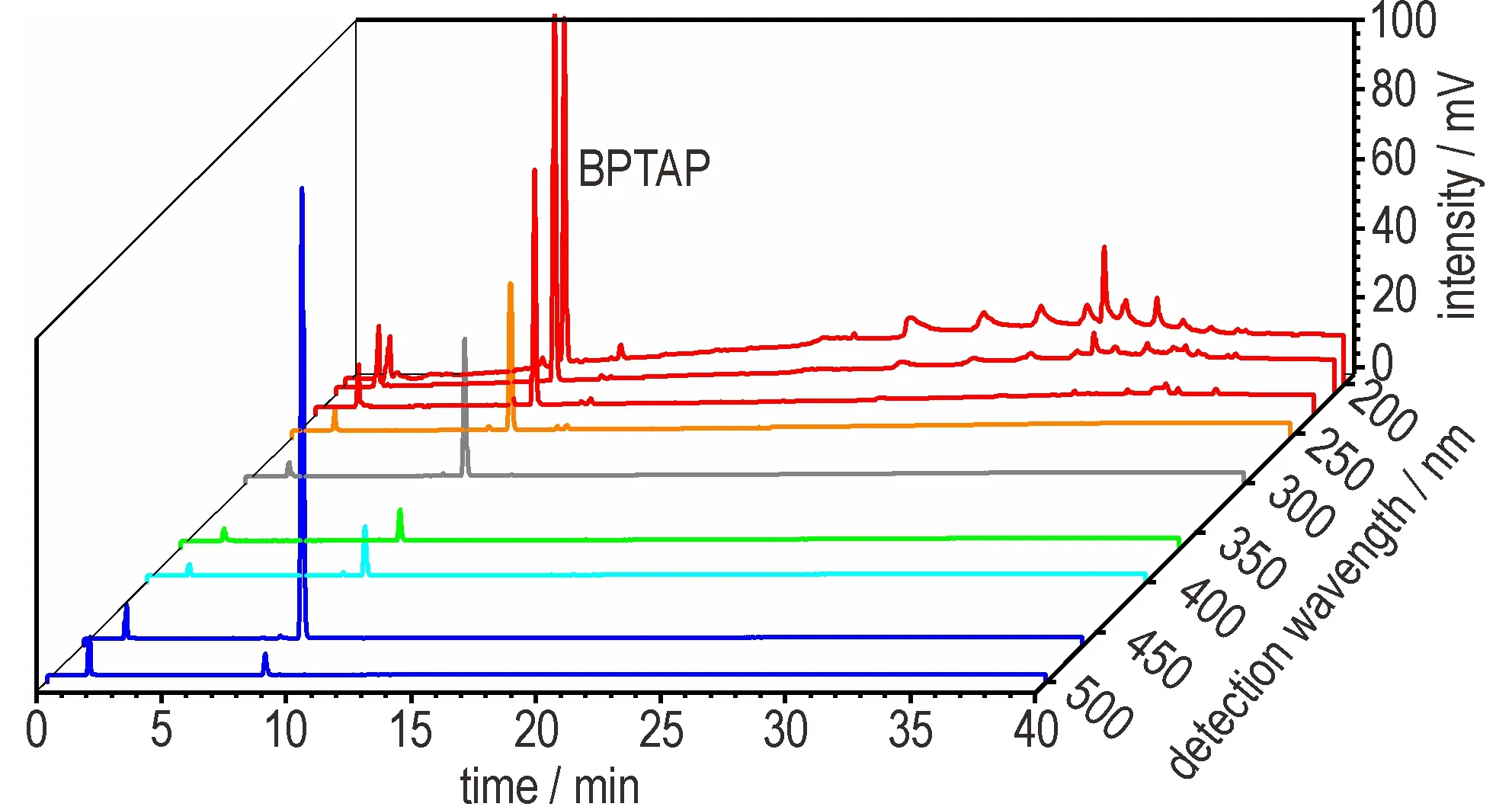
图6 不同波长下BPTAP 的色谱图Fig.6 Chromatographic analysis of BPTAP by using different detection wavelengths
3.1.6 梯度分离时间的优化
在梯度洗脱程序中,梯度分离时间不仅影响着样品的分离效果,也决定了分析的效率。本研究考察了不 同 的 梯 度 分 离 时 间(8、10、15、20 和25 min)对BPTAP 色谱分离的影响。由图7a 可知,梯度分离时间的缩短,分析效率也有所提升,且BPTAP 的峰高和峰面积均随之增加。然而,当梯度分离时间为8 min 时,BPTAP 与其相邻的色谱峰(保留时间为8.25 min)分离较差,未达到基线分离。因此,选择10 min 作为梯度分离的最佳运行时间。
3.1.6 进样量的优化
增大进样量可以有效的提高色谱的峰高和峰面积,但过大的进样量会造成色谱柱饱和,进而影响分离效果。如图7b 所示,为不同进样量(5、10、20、30 和40 μL)下BPTAP 的分离情况。当进样量为5 μL 时,色谱峰1、2 的峰面积较弱,甚至小于1000 mV·min,难以进行准确的定量分析。进样量为10 μL 时,色谱峰1、2的峰面积增加,均大于1500 mV·min,且分离效果良好,对称性高。当进样量为20~40 μL时,保留时间为0~4 min处各峰未能分离,色谱峰2的对称性下降。当进样量为40 μL 时,色谱峰2 与BPTAP 重叠,难以区分。因此,选择10 μL为BPTAP色谱分析的最佳进样量。

图7 梯度洗脱不同分离时间(a)和不同进样量(b)下BPTAP 的色谱分析Fig.7 Chromatographic analysis of BPTAP by using different separation time(a),and different injection volumes(b)
3.1.7 流速的优化
适当的流速能有效地减小被分析物的保留时间,节约时间成本的同时提高分析效率。本研究考察了流速分别为0.6、0.8、1.0、1.2 和1.5 mL·min-1时BPTAP的色谱分析效果(图8a)。随着流速的增加,BPTAP 的保留时间明显的缩短,由0.6 mL·min-1流速时的13.37 min 下 降 到1.5 mL·min-1流 速 时 的6.84 min。与此同时,BPTAP 的峰高(113507~69892 mV)和峰面积(917211~354022 mV·min)也随流速的增加而下降。当流速为1.0 mL·min-1时,BPTAP 具有合适的峰高(95258 mV)和峰面积(546268 mV·min),保留时间为9.16 min,且各峰的分离度良好(均大于1.95),基线平稳,信噪比低。因此,选择1.0 mL·min-1作为BPTAP 色谱分析的检测流速。
3.1.8 柱温的优化
为避免色谱柱的温度对BPTAP 的色谱分析产生不利影响,本研究考察了柱温箱分别为25、30、35、40 ℃和45 ℃时的色谱分析效果。结果如图8b 所示,柱温的升高会略微降低BPTAP 的保留时间。保留时间由25 ℃时的8.80 min 降至45 ℃的8.04 min,对应峰高由576509 mV 下降到545171 mV,相应峰面积由100381 mV·min下降到96671 mV·min。参考实验室环境温度,柱温为30 ℃时,BPTAP的保留时间为8.65 min,峰面积为572358 mV·min,峰高为100139 mV,且对称性好、柱效高。因此,最终选择30 ℃作为BPTAP 色谱分析的柱温。

图8 不同流速(a)和不同柱温(b)下BPTAP 的色谱分析Fig.8 Chromatographic analysis of BPTAP by using different flow rates(a)and column temperatures(b)
3.1.9 最佳的色谱分析条件
经上述优化,本文建立了基于HPLC 的BPTAP 纯度分析方法,并获得了最佳的色谱分析条件,其色谱柱为Plus C18 色谱柱(4.6×150 mm,5.0 μm),流动相为乙腈-水混合液(含0.1 mg·min-1的乙酸铵),梯度分离模式,乙腈含量在10 min内从35%线性增至90%并保持5 min,进样量为10.0 μL,柱温为30 ℃,流速为1.0 mL·min-1,检测波长为230 nm。最佳条件下BPTAP 的色谱图如图9a 所示,BPTAP 的保留时间为9.13 min,保留因子(K')为5.85,拖尾因子(T)为1.22,半峰宽(FWHM)为0.18,各个峰的分离度(R)均大于1.90,展现了良好的分离效果。BPTAP及其杂质的分离参数如表1所示。

表1 优化的HPLC 条件下BPTAP 及杂质的分离参数Table 1 The separation parameters of BPTAP and the impurities under optimum condition of HPLC
3.2 BPTAP 方法验证
3.2.1 标准曲线与灵敏度
为建立BPTAP 定量分析的标准工作曲线,将BPTAP 参比溶液逐级稀释,配制成201.20、100.60、50.30、10.06、5.03、1.01 μg·mL-1和0.50 μg·mL-1的标准工作溶液,使用外标法结合峰面积定量,获得BPTAP 的标准工作曲线(见图9b)。在0.5~200 μg·mL-1的范围内,BPTAP 的浓度与色谱峰面积呈现良好的线性关系,其线性方程为Y=33455.72X-45404.95,相关系 数R2=0.9997,检 测 限 为0.02 μg·mL-1,定 量 限 为0.07 μg·mL-1,表明该方法具有良好的灵敏度。

图9 优化HPLC 条件下BPTAP 的色谱图(a)及BPTAP 的标准工作曲线(b)Fig.9 The chromatogram of BPTAP under optimum condition of HPLC(a)and the standard curve for BPTAP(b)
3.2.2 准确度
在待测的BPTAP 样品中,分别加入50.30、100.60、201.20 μg·mL-1不同浓度的纯度参比溶液,在最佳HPLC 条件下,每个样品重复测定3 次,记录峰面积并计算加标回收率。如表2 所示,BPTAP 的加标回收率在101.68%~104.58%之间,且RSD 在0.19%~0.28%之间,说明本方法具有良好的准确性。

表2 BPTAP 在不同浓度下的加标回收率(n=3)Table 2 The recovery of BPTAP after adding different standard solutions
3.2.3 精密度
日间/日内精密度是分别对同一批样品在连续不同天数/同一天进行测试,用于表示仪器的环境条件发生微小变化时测定结果的差异性。为考察BPTAP 的HPLC 分析方法的精密度,同一样品分别制备6 份供试品,分别进样,连续测定3d,并以BPTAP 峰面积的RSD作为日间/日内精密度的评价依据。由表3 可知,BPTAP 峰 面 积 的 日 内RSD 均≦1.56%,日 间RSD 为1.17%,表明本方法的精密度良好。

表3 日间/日内精密度实验Table 3 The inter-day and intra-day precision
3.2.4 重复性
基于已建立的HPLC 分析方法,将BPTAP 配制成20 μg·mL-1的溶液,重复分析6 次,以考察BPTAP 色谱方法的重复性。如表4 所示,BPTAP 的保留时间、峰面积的相对标准偏差(RSD)分别为0.04%、0.81%,表明BPTAP 分析方法具有良好的重复性。

表4 BPTAP 色谱方法的重复性Table 4 The repeatability of BPTAP method
3.2.5 稳定性
使用新配制的20 μg·mL-1的BPTAP 溶液,分别在1 h、12 h、24 h 和48 h 等不同时间段内测量BPTAP 的保留时间和峰面积,以考察本方法对BPTAP 分析的稳定性。如表5 所示,BPTAP 在48 h 内保留时间、峰面积的RSD 分别为0.20%、0.93%。可见,基于HPLC 的BPTAP 分析方法在48 h 内是相对稳定的。

表5 基于HPLC 的BPTAP 分析方法的稳定性Table 5 The stability of BPTAP method base on HPLC
3.2.6 耐用性
实际实验过程中存在一些影响BPTAP 色谱分析的细微变化,如检测波长、色谱柱温的波动。为评估某些因素发生波动时对分析结果的影响,对这些因素进行细微调整如柱温(±2 ℃)、检测波长(±2 nm),考察变动因素对检测结果的影响。由表6 可知,在柱温(±2 ℃)和检测波长(±2 nm)的细微调整下,保留时间(tR)、分离度(R)和半峰宽(FWHM)均保持良好。可见,本方法具有优良的耐用性,色谱条件的细微波动不影响分离效果。

表6 BPTAP 的HPLC 方法在不同条件下的耐用性Table 6 The robustness of BPTAP method under different conditions
3.3 实际样品分析
为考察本方法在BPTAP 中的实际应用前景,还将本方法应用于BPTAP 实际样品重结晶过程中的纯度分析研究。如表7 所示,不同批次(20190601 批、20190917 批、20191015 批和20191119 批)的BPTAP样品的纯度存在差异,粗品纯度最低为80.37%,经过一次重结晶后纯度提升至94.32%,二次重结晶后的纯度提升至95.47%。结果表明,本研究建立的BPTAP 色谱分析方法适合用于BPTAP 的纯度分析研究。

表7 不同批次的BPTAP 和重结晶之后的纯度分析Table 7 The purity analysis of BPTAP in different batches and after the recrystallization
4 结论
针对BPTAP 研制生产及应用过程中对纯度分析的需求,开展了基于高效液相色谱(HPLC)的分析方法构建、验证及实际应用研究。
(1)首先,通过优化影响色谱分析的相关因素如色谱柱、进样量及分析时间等,建立了基于HPLC 的BPTAP 纯度分析方法。基于该方法,BPTAP 及其杂质得到高效分离,BPTAP 的保留时间为9.13 min,各个峰的分离度均大于1.9。
(2)接着,对该分析方法进行了系统验证,该方法在0.5~200 μg·mL-1范围内展现出良好的线性关系,检测限为0.02 μg·mL-1,定量限为0.07 μg·mL-1,并体现了良好的精密度和重现性。
(3)此外,还将该方法应用于BPTAP 实际样品重结晶过程的纯度监测研究,分析发现不同批次的BPTAP 样品存在纯度差异,且经过重结晶可以有效提高BPTAP 的纯度。
总之,本研究建立的BPTAP 色谱分析方法具有准确、高效、灵敏等优势,可用于BPTAP 的纯度分析、含量检测和质量控制等研究。
