头孢氨苄胶囊在健康受试者中的生物等效性和药动学/药效学研究
2021-07-21胡盈盈郁继诚陈渊成戴静怡王晶晶武晓捷程洁如丁玲玲刘晓雪龚玉秀
金 逸,胡盈盈,郁继诚,陈渊成,戴静怡,王晶晶,张 菁,武晓捷,程洁如,丁玲玲,刘晓雪,龚玉秀
头孢氨苄为第一代头孢菌素类口服抗生素,于20世纪60年代应用于临床,为目前使用最广、最多的一类抗生素。其抗菌谱与头孢噻吩相仿,但其抗菌活性较后者为差。除肠球菌属和甲氧西林耐药葡萄球菌外,肺炎链球菌、溶血链球菌、产或不产青霉素酶葡萄球菌的大部分菌株对头孢氨苄敏感。头孢氨苄对奈瑟菌属有较好抗菌作用,但对流感嗜血杆菌的作用较差,对部分大肠埃希菌、奇异变形杆菌、沙门菌和志贺菌有一定抗菌作用。其他肠杆菌科细菌、不动杆菌、铜绿假单胞菌、脆弱拟杆菌对头孢氨苄均耐药,梭杆菌属和韦荣球菌一般敏感,厌氧革兰阳性球菌中度敏感。因其临床广泛应用,上海新亚药业闵行有限公司研发了国产头孢氨苄胶囊,并研究比较国产头孢氨苄胶囊(受试制剂)和原研产品武田药品工业株式会社生产的头孢氨苄胶囊(商品名为Sencephalin®)(参比制剂)的生物等效性,遵循我国《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究指导原则》[1]及《药物临床试验质量管理规范(2020年版)》[2],于2018年9月在复旦大学附属华山医院Ⅰ期临床研究中心开展生物等效性临床试验。由于药动学/药效学(PK/PD)研究可以探讨量效关系和时效关系,使感染病灶维持有效的药物浓度,保证药物起效且不产生毒性作用和不良反应,实现给药方案优化。因此,本文还同时基于PK数据开展PK/PD研究,评价说明书所载给药方案及其优化策略,为该药临床合理应用提供依据。
1 材料与方法
1.1 研究设计
本研究采用单中心、随机、开放、单剂量、两周期、两交叉、空腹或餐后给药试验设计。入选研究的受试者按照1∶1的比例随机进入(空腹与餐后)2个试验组,每组受试者分别按照方案规定先后给予受试制剂-参比制剂、参比制剂-受试制剂,两周期自身交叉,空腹给药或高脂餐后给药,并按设计的时间点采集血样,两周期间清洗期为7 d。本临床试验获得了复旦大学附属华山医院伦理委员会批准。药品审评中心备案号为B201800242-02。
1.2 受试者
所有入组受试者均签署知情同意书。受试者无临床严重疾病史或循环系统、内分泌系统、神经系统疾病或血液系统、免疫系统、精神疾病及代谢异常等病史,无消化不良、胃炎、食管炎、十二指肠溃疡或胃溃疡等胃肠道疾病史以及胃肠道手术史,无对头孢菌素类药物及辅料(玉米淀粉、乳糖、甲基纤维素、硬脂酸镁、明胶空心胶囊)中任何成份过敏者、无青霉素过敏史者,无过敏体质,无食物、药物过敏史者。人类免疫缺陷病毒抗体、丙型肝炎抗体、乙型肝炎病毒表面抗原、梅毒检查结果阴性,体格检查、实验室检查(血常规、尿常规、凝血检查、血液生化检查、血妊娠)正常或异常无临床意义,且内生肌酐清除率<90 mL/min,心电图及生命体征正常,筛选前两周内未服用过任何药品(包括非处方药、中草药及维生素),酒精呼吸检查和烟检阴性,试验期间统一饮食,禁止吸烟、饮酒、摄入饮料,禁止剧烈运动。
1.3 试验用药品和给药方式
受试制剂头孢氨苄胶囊,规格:0.125g/粒,有效期:2019年11月,批号:9400101,上海新亚药业闵行有限公司生产。参比制剂头孢氨苄胶囊,规格:0.125 g/粒,有效期:2020年4月,批号:HJ1014,武田药品工业株式会社生产。
共入组56名受试者,每周期服药前禁食至少10 h。空腹组中受试制剂组受试者于服药当天早上8点左右按照随机表单剂量口服受试制剂1粒(0.125 g),用240 mL温水送服;参比制剂组受试者单剂量口服参比制剂1粒(0.125 g),用240 mL温水送服。餐后组中受试者于服药当天早上07:30左右进食高脂餐(高脂:脂肪在提供的食物中约占50%的热量;高热:约3348.8~4186.0 kJ饮食。其中蛋白质约提供627.9 kJ热量,碳水化合物约提供1046.5 kJ热量,脂肪约提供2093.0~2511.6 kJ热量),开始用餐30 min后单剂量口服受试制剂1粒(0.125 g),用240 mL温水送服;参比制剂组受试者口服参比制剂1粒(0.125 g),用240 mL温水送服。服药前1 h及服药后2 h内禁止饮水,此后2 h内可以饮水量≤100 mL,之后每2 h可以饮水量≤200 mL,直至采血结束,10 h内饮水量≤900 mL。服药4 h后进食午餐(统一标准饮食)。第一周期受试者服用受试制剂或参比制剂,则第二周期服用参比制剂或受试制剂,流程与第一周期相同。
1.4 血样采集及处理
给药前在受试者前臂静脉埋置留置针头,每周期于给药前0 h(给药前3 h内,视为0 h)和给药 后10 min、20 min、35 min、45 min、1 h、1 h 20 min、1 h 40 min、2 h、2 h 30 min、3 h、4 h、5 h、6 h、8 h和10 h共16个时间点采集静脉血4 mL置于预冷且已标记好的含EDTA-K2抗凝剂真空采血管中,轻柔颠倒混匀,采集后即刻置于冰浴中,采血后30 min内于2~8℃、约1 850×g条件下离心10 min,分离出血浆,0.8 mL分装于检测管中,剩余量分装于备份管,存入-60℃以下低温冰箱中,直至样本转运至生物样本检测单位。从血样采集到血浆样品放置到冰箱的过程不能超过1 h,均为黄光灯冰浴下操作。
1.5 测定方法
血浆药物浓度测定采用液质色谱-质谱联用法。
1.5.1 测定条件 色谱柱:SHIMADZU, Shimpack GISS-HPC18(2.1×50 mm);流动相A:含0.1%甲酸(FA)的水溶液,流动相B:含0.1%FA的100%甲醇溶液。柱温40℃;流速:0.4 mL/min,自动进样器温度设置为4℃,进样量2 μL。头孢氨苄离子对:m/z 348.15→m/z 174.20,内标离子对:m/z 353.10→m/z 179.10。
1.5.2 血浆样本前处理 样品在湿冰和黄光灯下融化后,涡旋混匀。吸取50 μL样品至96孔板中,对于双空白样品或QC样品,加入50 μL空白基质。加入50 μL内标溶液,对于双空白样品加入50 μL 0.1% FA的50%甲醇溶液。加入100 μL的10%三氯乙酸的50%甲醇溶液,然后涡旋约10 min,在约4℃条件下2 000×g离心10 min。转移20 μL上清溶液至已加入280 μL 0.1% FA的50%甲醇溶液的96孔板中,涡旋5 min,待进样。
1.6 方法学验证
标准曲线浓度水平分别为:20.0、40.0、160.0、640.0、1 600.0、6 400.0、8 000.0和10 000.0 μg/L。检测得到头孢氨苄峰面积与内标峰面积之比和浓度线性回归方程为:y=0.764302x-0.000683,R2>0.985。线性范围20.0~10 000.0 μg/L,定量下限为20.0 μg/L。在测定条件下,待测物头孢氨苄和内标保留时间均约为0.2 min,头孢氨苄和内标保留处峰面积未见明显干扰,未见明显残留。精密度和准确度采用3个分析批5个浓度水平[极低浓度、低浓度、中浓度(几何均值)、中浓度和高浓度](LLOQC、LQC、GMQC、MQC、HQC)的质控样品,每个浓度水平6个平行。批内相对误差百分率平均在-2.6~3.3,变异系数在1.5~6.4。批间相对误差百分率平均在-1.2~1.9,变异系数在2.8~5.8。HQC、MQC、LQC血浆质控样品的提取回收率分别为97.0%、94.5%、94.0%,变异系数为1.7。内标平均提取回收率约为101.5%,变异系数为2.7。低、高浓度下,LQC、HQC浓度质控内标归一化基质效应因子变异系数分别为2.0、1.0。脂血、溶血基质对待测物和内标测定无显著影响。待测物储备液和工作溶液在-20℃条件下24 d稳定,在湿冰黄光条件下分别为26、25 h稳定;全血在湿冰条件下,黄光灯或日光灯条件下均2 h稳定;血浆基质样品室温黄光灯与湿冰日光灯条件下,均2 h稳定,湿冰黄光灯下21 h稳定,-20℃条件下40 h稳定,-60℃条件下62 d稳定;处理后样品在4℃条件下42 h稳定;血浆基质样品在-60℃条件下4次反复冻融后稳定。
1.7 PK参数计算及生物等效性判定
采用Phoenix WinNonlin软件(版本8.1,美国Pharsight公司)根据实际的采血时间点进行非房室模型PK参数计算。对主要PK参数(Cmax、AUC0-t、AUC0-∞)进行自然对数转换后进行分析,对Tmax进行非参数检验。以方差分析结果为基础,采用90%置信区间分析法,当受试制剂与参比制剂PK参数AUC0-t、AUC0-∞和Cmax的几何均值比的90%置信区间均在80.00%~125.00%等效区间内,则认为两制剂生物等效。
1.8 PK/PD分析
1.8.1 PK/PD指数计算 对头孢氨苄个体PK数据建立房室模型,模型结构包括一室模型或二室模型,吸收分为延迟或无延迟,权重从1、1/C或1/C2中选取。药品说明书所载的给药方案为:250~500 mg、1次/6 h,高剂量4 g/d,本研究针对说明书中给药方案开展PK/PD研究。基于个体房室模型PK参数模拟头孢氨苄多次给药的PK特征,并计算头孢氨苄多次给药达稳态时浓度高于MIC时间占给药间隔的百分比(%T>MIC),公式为(t2-t1)/τ×100%,其中t1和t2分别为MIC与药时曲线相交的左边时间点和右边时间点。MIC为最低抑菌浓度,取值0.031 25、0.062 5、0.125、0.25、0.5、1、2、4、8、16和32 mg/L。采用Matlab软件(R2014a, 美国Mathworks公司)开展本部分工作。
1.8.2 蒙特卡洛模拟 根据文献报道[2],①设头孢氨苄%T>MIC靶值为40%;②计算MIC取特定数值时头孢氨苄空腹给药%T>MIC达到靶值的概率(PTA): 各房室模型参数按对数正态分布产生随机数(2 000个),采用Matlab软件计算%T>MIC并统计超过该靶值个数占总体的百分比。
1.9 结果表示和统计学分析
采用SPSS软件22.0版(美国IBM公司)开展统计学检验,P<0.05为差异具有统计学意义。
2 结果
2.1 受试者情况
空腹组28名受试者和餐后组28名受试者全部完成试验。空腹组平均年龄(28.7±5.3)岁,平均体重(61.16±6.47) kg,平均体重指数(22.46±1.85)kg/m2。餐后组平均年龄(28.7±6.1)岁,平均体重(61.40±7.94) kg,平均体重指数(22.17±1.93) kg/m2。
2.2 头孢氨苄PK
头孢氨苄胶囊空腹组受试者血药浓度结果见图1,餐后组试验受试者血药浓度结果见图2。无论空腹或餐后给药,服用受试制剂和参比制剂后药时曲线均相似。
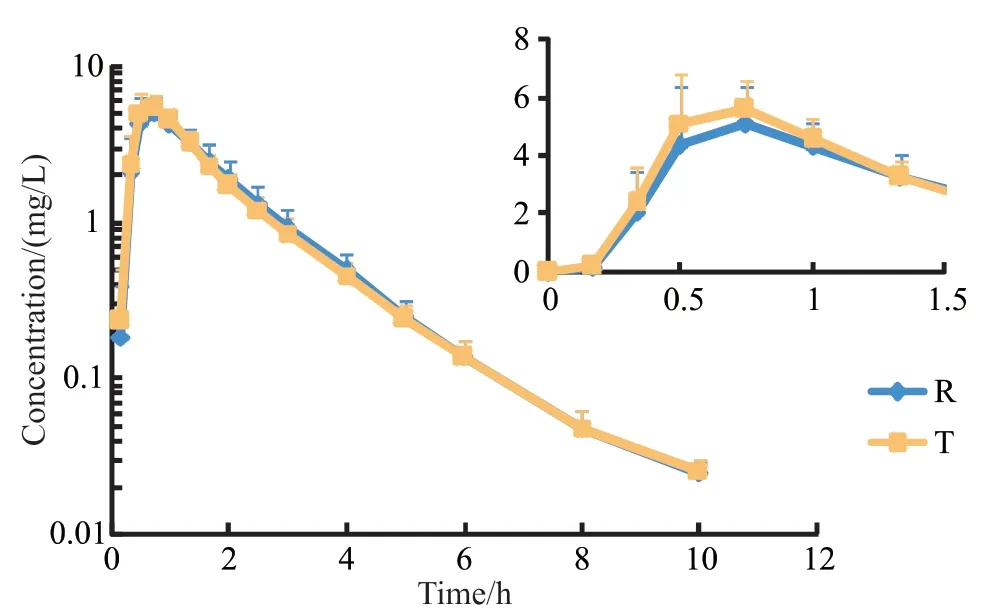
图1 28名受试者空腹口服头孢氨苄后平均血药浓度-时间曲线Figure 1 The mean concentration - time curves after oral administration of cefalexin under fasted condition in 28 healthy subjects

图2 28名受试者餐后口服头孢氨苄后平均血药浓度-时间曲线Figure 2 The mean concentration - time curves after oral administration of cefalexin under fed condition in healthy subjects
空腹和餐后口服头孢氨苄受试制剂和参比制剂后主要PK参数见表1。空腹组参比制剂和受试制剂给药后吸收迅速,于给药后0.73 h(中位数)达峰,两制剂Cmax和AUC相近,消除半衰期均较短,约为1.3 h。
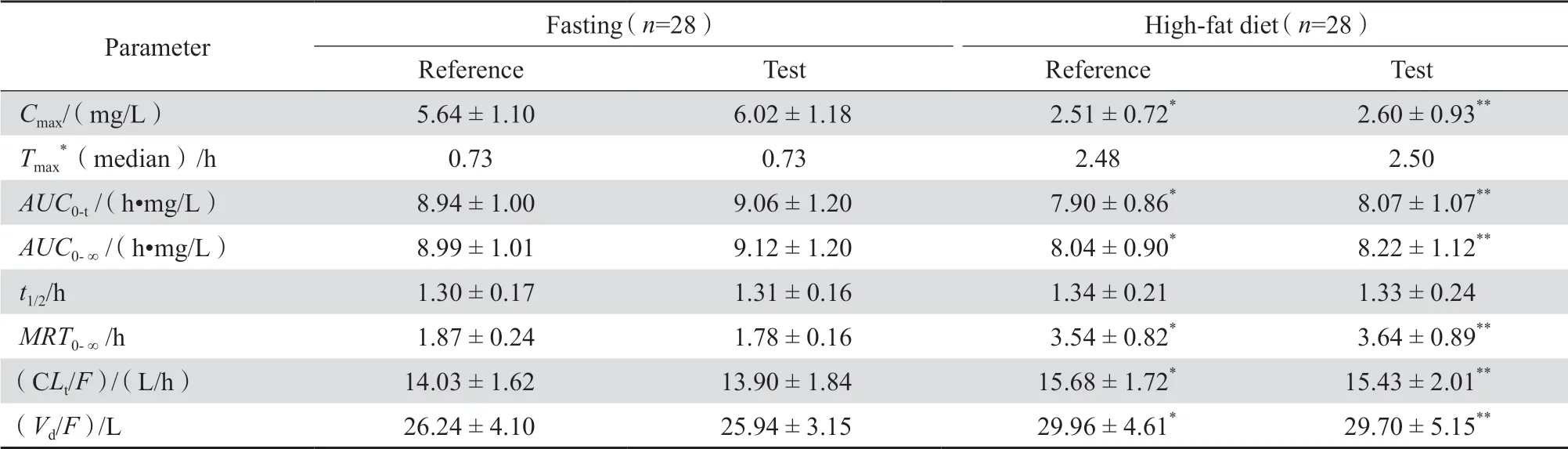
表1 空腹或高脂餐后口服头孢氨苄胶囊受试制剂和参比制剂主要药动学参数 Table 1 Pharmacokinetic parameters of cefalexin capsules and reference preparation after oral administration under fasting or high-fat diet condition
进高脂餐后口服头孢氨苄较空腹给药,受试制剂和参比制剂吸收均延迟,达峰时间约为2.50 h,Cmax均降低至一半左右,但AUC与空腹组相似。
头孢氨苄空腹给药时的PK符合吸收有延迟的二房室模型。对于受试制剂,头孢氨苄吸收速率(ka)和吸收延迟时间(Tlag)分别为(3.05±1.09)L/h和(0.27±0.03) h,中央室清除率(CL)和中央室-外周室之间清除率(Q)分别为(14.23±1.92) L/h和(9.55±5.43) L/h,中央室和外周室分布容积分别为(8.71±2.59)L和(7.65±2.15)L。
2.3 生物等效性评价
空腹口服受试制剂和参比制剂后,主要PK参数Cmax、AUC0-∞、AUC0-t几何均值比(受试制剂/参比制剂)分别为106.91%、101.22%、101.16%,90%置 信 区 间 分 别 为101.45%~112.66%、99.14%~103.34%、99.07%~103.29%;经非参数秩和检验,Tmax差异无统计学意义。高脂餐后服用受试制剂和参比制剂后,主要PK参数Cmax、AUC0-∞、AUC0-t的 几 何 均 值 比 分 别 为102.29%、102.08%、101.83%,90%置信区间分 别 为94.41%~110.84%、100.24%~103.95%、99.86%~103.84%;经非参数秩和检验,Tmax差异无统计学意义。空腹或餐后口服受试制剂和参比制剂后,Cmax、AUC0-∞、AUC0-t的几何均值比值(受试制剂/参比制剂)90%置信区间均落在80.00%~125.00%范围内,因此两制剂生物等效。
2.4 安全性评价
空腹状态下服用受试制剂的28例受试者共发生6例(6/28,21.4%)9例次的不良事件,服用参比制剂的28例受试者共发生7例(7/28,25.0%)12例次的不良事件。餐后状态下服用受试制剂的28例受试者共发生9例(9/28,32.1%)16例次的不良事件,服用参比制剂的28例受试者共发生4例(4/28,14.3%)9例次的不良事件。空腹组与餐后组所有不良事件均为1级(轻度),呈一过性,自行恢复,均与研究药物无关。受试制剂和参比制剂单剂给药后安全性好。
2.5 PK/PD分析
多剂空腹给予头孢氨苄达稳态时%T>MIC见图3。MIC=0.5 mg/L时,125、250、500和1 000 mg(均为1次/6 h)给药方案%T>MIC均值分别为60%、79%、96%和100%;MIC=1 mg/L时,四种给药方案%T>MIC分别为41%、60%、79%和96%。
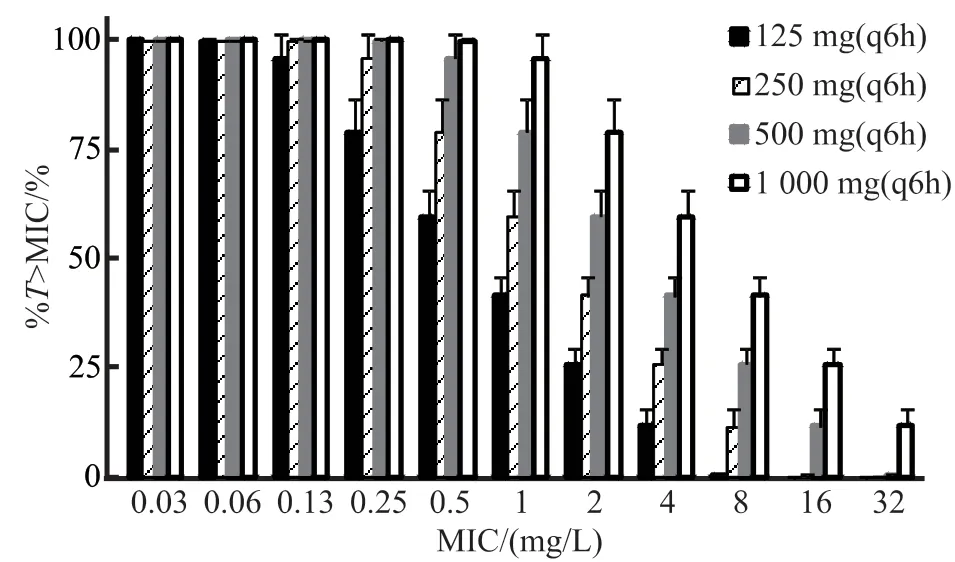
图3 多剂空腹给予头孢氨苄受试制剂达稳态时的%T>MICFigure 3 The %T>MIC of cefalexin (test preparation) at steady state following multiple oral doses under fasted condition
多剂空腹口服头孢氨苄不同剂量250、500、1 000 mg 1次/6 h达 稳 态 时%T>MIC的PTA见图4。随着头孢氨苄剂量增加,PTA出现不同程度增加。当对致病菌MIC≤0.5 mg/L时,250 mg(1次/6 h)的给药方案PTA超过90%。当对致病菌MIC≤1 mg/L时,500 mg(1次/6 h)给 药 方案PTA超过90%。当对致病菌MIC≤2 mg/L时,1 000 mg (1次/6 h)给药方案PTA超过90%。因此当PTA≥90%时,对致病菌三种给药方案可覆盖MIC值分别为0.5、1和2 mg/L。
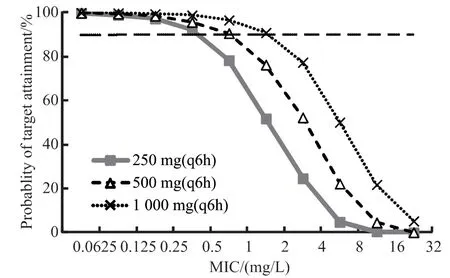
图4 多剂空腹口服头孢氨苄达稳态时%T>MIC的达标概率Figure 4 Probablity of target attainment of %T>MIC at steady state following multiple oral doses of cefalexin (test preparation) under fasted condition
3 讨论
临床试验中关于样本操作要求全程避光,故从采血至样本存入冰箱均使用黄光灯。但据文献报道[3],头孢氨苄光照10 d试验,各项指标无显著变化,都在合格范围内,表明头孢氨苄对光较为稳定。因此,后续的头孢氨苄研究中无须采取避光措施。
本研究结果显示,空腹给药头孢氨苄吸收较快,血药浓度在2 h内可达最高值。鉴于空腹和餐后达峰时间不同,空腹时在体内达峰时间短而进餐后则延迟,因此本研究兼顾两者进行了采血时间设置,在1 h前增加采血点,在2 h后也增加了2.5 h、3 h,使结果更易分析和评估。
从血药浓度-时间曲线图可以明显看出进餐对头孢氨苄PK存在影响:吸收速率明显下降。空腹口服头孢氨苄,吸收速率较快且达峰时间短;餐后口服头孢氨苄,吸收速率较慢且达峰时间延长。
按剂量校正后,本试验所得头孢氨苄Cmax和AUC与文献[4]接近,包括空腹和餐后。此外,本研究所得Cmax还与文献[5]接近,但AUC低于文献[5]报道结果。
PK/PD研究显示,头孢氨苄250、500、1 000 mg(1次/6 h)给药方案,对于MIC≤0.5、1、2 mg/L致病菌引起的感染,%T>MIC达标概率高于90%。因此临床应用头孢氨苄治疗时,可根据药敏结果采用不同的给药方案,实现个体化给药策略。本试验餐后组进食高脂餐,不是普通餐,因而该组PK研究结果不能代表进普通餐后头孢氨苄PK,因而未开展餐后组PK/PD分析。
头孢氨苄受试制剂和参比制剂吸收速度/程度以及消除均接近,PK具有生物等效性。对于MIC≤0.5 mg/L细菌引起的感染,采用头孢氨苄250~500 mg 1次/6 h治疗方案可预期取得较好微生物学疗效。
