Dispersive liquid-liquid microextraction,an effective tool for the determination of synthetic cannabinoids in oral fluid by liquid chromatography-tandem mass spectrometry
2021-07-20PierpoloTomiAlessndrGentiliRoertCuriniRossellGottrdo
Pierpolo Tomi,Alessndr Gentili,*,Roert Curini,Rossell Gottrdo,
Franco Tagliarob,Salvatore Fanalic
aDepartment of Chemistry,Sapienza University,P.le Aldo Moro 5,00185,Rome,Italy
bDepartment of Diagnostics and Public Health,University of Verona,Verona,Italy and Institute of Translational Medicine and Biotechnology,Sechenov First Moscow State Medical University,Moscow,Russia
cTeaching Committee of Ph.D.School in Natural Science and Engineering University of Verona,Verona,Italy
Keywords:
Microextraction techniques
Dispersive liquid-liquid microextraction
Illicit drugs
Synthetic cannabinoids
Silica C-based column
A B S T R A C T
In the present work,dispersive liquid-liquid microextraction(DLLME)was used to extract six synthetic cannabinoids(JWH-018,JWH-019,JWH-073,JWH-200,or WIN 55,225,JWH-250,and AM-694)from oral fluids.A rapid baseline separation of the analytes was achieved on a bidentate octadecyl silica hydride phase(Cogent Bidentate C18;4.6 mm × 250 mm,4 μm)maintained at 37 °C,by eluting in isocratic conditions(water:acetonitrile(25:75,V/V)).Detection was performed using positive electrospray ionization-tandem mass spectrometry.The parameters affecting DLLME(pH and ionic strength of the aqueous phase,type and volume of the extractant and dispersive solvent,vortex and centrifugation time)were optimized for maximizing yields.In particular,using 0.5 mL of oral fluid,acetonitrile(1 mL),was identified as the best option,both as a solvent to precipitate proteins and as a dispersing solvent in the DLLME procedure.To select an extraction solvent,a low transition temperature mixture(LTTM;composed of sesamol and chlorine chloride with a molar ratio of 1:3)and dichloromethane were compared;the latter(100μL)was proved to be a better extractant,with recoveries ranging from 73% to 101% by vortexing for 2 min.The method was validated according to the guidelines of food and Drug Administration bioanalytical methods:intra-day and inter-day precisions ranged between 4% and 18% depending on the spike level and analyte;limits of detection spanned from 2 to 18 ng/mL;matrixmatched calibration curves were characterized by determination coefficients greater than 0.9914.Finally,the extraction procedure was compared with previous methods and with innovative techniques,presenting superior reliability,rapidity,simplicity,inexpensiveness,and efficiency.
1.Introduction
Synthetic cannabinoids(SCs)include a considerably large family of compounds,but only a few of them are structurally related to the primary psychoactive component of cannabis,Δ9-tetrahydrocannabinol(Δ9-THC)[1].Most SCs are 3-indole derivatives,usually referred to by trade names related to their devisers,e.g.,JWH-XXX(John W Huffmann)[2],CP-XX,XXX(Charles Pfizer),HU-XXX(Hebrew University),and AM-XXXX(Alexandros Makriyannis).Within a trade name group,several chemical subgroups are included,such as benzoyl,naphthol,phenylacetyl,alkyl,piperazinyl,carboxylate,carboxamide,thiazolyl,and naphthylmethyl derivatives[1,3].In this context,the JWH series represents one of the main SC subgroups detected in herbal products,with brand names such as “Spice”, “K2”, “herbal incense”, “Cloud 9”,and“Mojo”[4].These apparently innocuous articles are sold on the Internet or in convenience stores disguised as products to perfume the environment or for other unoffending purposes.The direct consequence of this easy availability is the widespread abuse of such drugs,especially among younger individuals in whom an increasing number of intoxication cases have been recorded.To curb the circulation of these products,control institutions need quick,simple,and reliable analytical methods to perform analyses of a large number of biological and seized specimens containing the target and/or unidentified SCs.To date,most analytical methodologies developed rely on gas chromatography and liquid chromatography coupled with mass spectrometry(GC-MS and LC-MS).In clinical and forensic toxicology,in addition to serum/blood[5-8]and herbal blends[9-12],methods of special interest are also being developed to screen and/or Confirm the occurrence of SCs in“noninvasive”biological matrices such as urine[13-16]and saliva[17-22],with the latter considered as one of the most accessible biological fluids.In particular,oral fluid analysis has proved to be a practical solution for the detection of SCs,especially during roadside or workplace drug testing.Moreover,saliva collection is a simple procedure that does not require the presence of medical staff.To date,methods analyzing SCs in the oral fluid have employed conventional extraction procedures based on liquidliquid extraction(LLE)or solid-phase extraction(SPE).Although valid,these approaches fail to adequately meet the high throughput requirements implemented owing to the need to monitor a significant number of samples. Dispersive liquid-liquid microextraction(DLLME)is a valid technique for the extraction of several classes of compounds from aqueous environmental samples[23,24];rapidity is one of its distinct merits.Nevertheless,its application in the treatment of biological fluids warrants the proper selection of a dispersing solvent,which should allow both dispersion of the extractant in the aqueous sample and precipitation of peptides and proteins.This aspect is nearly always neglected[25-27],and if not precisely addressed,it can be responsible for:i)an imperfect separation between the aqueous and organic phases after the DLLME centrifugation step,ii)difficulty in recovering the settled phase,and iii)stress on the chromatographic system.
In the present work,for the first time,a DLLME procedure was developed to extract SCs from oral fluids.The new extraction procedure was carefully evaluated and organized to overcome the above-mentioned analytical problems.Accordingly,special attention was paid to the two-fold role that the dispersing solvent should satisfy when DLLME is applied to biological fluids and,particularly,to saliva.High-performance liquid chromatography-tandem mass spectrometry(HPLC-MS)was used for analyte quantitative determination.Once optimized,the DLLME method was compared with conventional SPE,as well as innovative solutions(DLLME based on the use of a deep eutectic solvent as extraction solvent;rotating disc-SPE using buckypaper),to achieve the best results.Finally,this work incorporated innovative experiments on i)the chromatographic behavior of these analytes on a bidentate C18silica hydride phase in comparison with a conventional C18,and ii)the optimization of an isocratic separation,which is another important timesaving strategy in clinical or forensic routine investigations.
2.Materials and methods
2.1.Chemicals,materials,and solutions
The analytical standards of JWH-018,JWH-019,JWH-073,JWH-200,JWH-250,and AM-694 were obtained from LGC Standards(Sesto San Giovanni,Milan,Italy).The exact masses and log P of the analytes are shown in fig.1.Choline chloride and sesamol,with purity grade greater than 98%,methanol,ethanol,acetonitrile,ethyl acetate,tetrahydrofuran(THF),chloroform(RS-PLUS grade),and formic acid(50%,V/V)were purchased from Sigma-Aldrich.Commercial buckypaper(BP)was purchased from Nanolab,Inc.(Nanolab,Waltham,USA).Oasis HLB cartridges(500 mg,6 mL)were purchased from Waters Corporation(Milford,MA,USA).Ultrapure water was obtained using a Milli-Q water generator(Millipore,Bedford,MA,USA).Individual stock solutions were prepared in methanol at a concentration of 1 mg/mL.Working standard solutions were prepared by diluting the stock solutions with methanol at proper concentrations according to the various stages of the experimental work.All solutions were stored at-20°C.
2.2.Liquid chromatography-tandem mass spectrometry
The samples were analyzed with a PerkinElmer series 200 HPLC system equipped with an autosampler(PerkinElmer,Norwalk,CT).Chromatographic separation was performed on a Cogent Bidentate C18(4.6 mm×250 mm,4μm)column(MTC,Eatontown,NJ,USA)maintained at 37°C.The analysis time of a single run was 15 min using isocratic conditions:water:acetonitrile(25:75,V/V)at a flow rate of 1 mL/min.The mobile phase was split employing a postcolumn T-valve,leading to 200μL/min into the electrospray ionization(ESI)source of the mass spectrometer.After each injection,the autosampler needle was washed with acetonitrile.
The analytes were detected with a PE-Sciex API-3000®(PerkinElmer Sciex Toronto,Canada)triple quadrupole mass spectrometer.The ESI source was operated in positive ionization with a capillary voltage of+4500 V.High purity nitrogen was used both as curtain gas and collision gas,while air was employed as the nebulizer gas and drying gas.The latter was heated by adjusting the source heater temperature to 350°C.The full width at half maximum(FWHM)was set at m/z 0.7±0.1 in each mass-resolving quadrupole to operate with a unit resolution.The multiple reaction monitoring(MRM)mode was used for analyte quantification.Two MRM transitions were selected per analyte,for a total of 12 ion currents monitored with a pause time of 5 ms.All LC-MS parameters useful for identification and quantification are listed in Table 1.The LC-MS data were processed using Analyst®1.5 Software(AB Sciex).
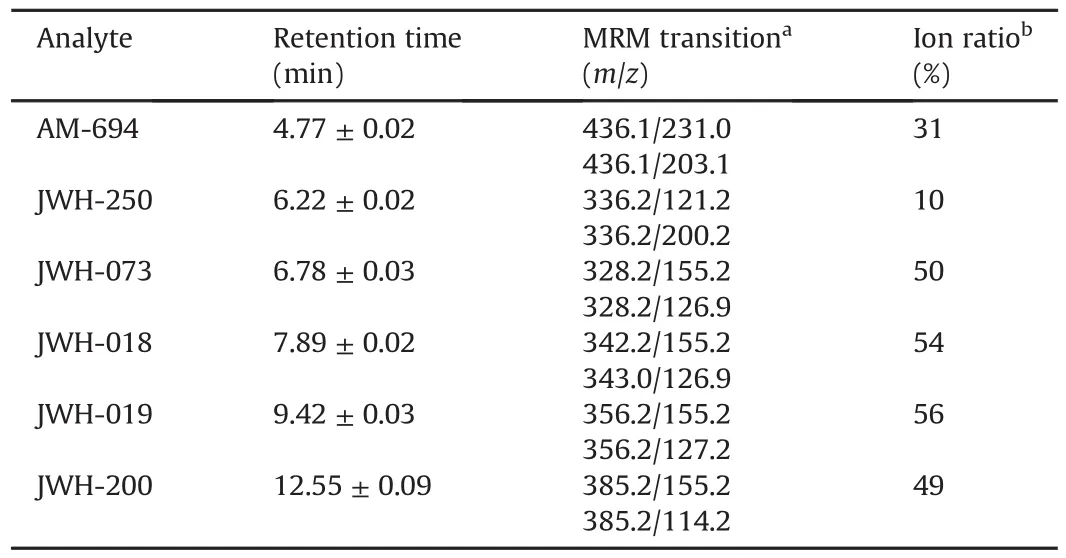
Table 1 LC-MS parameters used for the six synthetic cannabinoids(SCs)identification in oral fluid samples.Retention time and ion ratio were calculated as mean of three replicates.
2.3.Sample collection
For the optimization and validation of the DLLME method,oral fluid was collected from healthy volunteers(n=5)after obtaining consent.Each sample(approximately 1 mL)was directly collected into a 15-mL polypropylene tube,30 min after a meal or after tooth brushing to obtain residue-free samples.The saliva from different donors was pooled(20 mL)and used for method validation.The samples were maintained at-20°C until use.
2.4.Preparation of chlorine chloride(sesamol)3
For a low transition temperature mixture(LTTM)preparation,both choline chloride and sesamol were dried individually in an oven at 50°C for 24 h.The solid mixture of the two components was prepared by weighing 1.000 g of choline chloride and 1.979 g of sesamol to achieve a molar ratio of 1:3.Next,the solid mixture was heated at 50°C to favor the quick formation of an amber viscous liquid(4 mL).The LTTM was cooled and later stored at room temperature until use.For extraction,the LTTM was slightly heated to reduce its viscosity and favor its sampling using a microsyringe.
2.5.Sample preparation
The extraction procedure for SCs from oral fluid involved two steps:i)protein precipitation and ii)DLLME procedure.
2.5.1.Protein precipitation
An aliquot of 0.5 mL of oral fluid was placed into a 15 mL centrifuge tube,and 1 mL of ice-cold acetonitrile was added.The mixture was sonicated for 5 min to promote protein precipitation and centrifuged at 12,500 g for 5 min.The supernatant was transferred to a 15 mL centrifuge tube for the subsequent DLLME step.
2.5.2.DLLME procedure
The supernatant obtained in the previous step(about 1.5 mL)was diluted with 4.5 mL of water;acetonitrile(used for protein precipitation)acted as a dispersing solvent.Thus,100μL of chloroform(extraction solvent)was directly injected into the supernatant.The mixture was vigorously vortexed for 2 min to favor the formation of a cloudy solution.After centrifugation at 12,500 g for 5 min,the settled phase was isolated using a microsyringe,transferred into another tube,and evaporated to dryness under nitrogen flow in a water bath at 40°C.The residue was reconstituted with 50μL of water:methanol(25:75,V/V)and injected into the HPLCMS system(5μL).
2.6.Method validation
The DLLME-HPLC-MS method for the analysis of SCs in oral fluids was validated by assessing recovery,precision,linearity,limit of detection(LOD),and lower limit of quantitation(LLOQ).The most intense MRM transition(MRM1)was used for quantitative purposes and to calculate LOD and LLOQ,whereas the second most intense one(MRM2)was chosen for confirmation purposes.
When required,blank samples were spiked pre-extraction with the analytes and vortexed for 5 min to favor their mixing with the matrix.Recoveries and precisions(intra-day and inter-day)were evaluated at the LLOQ and 10 times the LLOQ,analyzing five replicates for each spike level on the same day.Inter-day precision was evaluated by analyzing 10 replicates over a two-week period.
Matrix-matched calibration curves were built by spiking six blank aliquots with increasing concentrations of the analytes(0.00,0.03,1,10,20,35,and 50 ng/mL)pre-extraction.The curves were constructed by linear regression,plotting the peak area against the spike level.
LOD and LLOQ were experimentally estimated as the minimum concentration of an analyte in oral fluid capable of providing a signal-to-noise ratio of 3 and 5,respectively.
3.Results and discussion
3.1.Optimization of the chromatographic conditions
Based on the chemical structure of the evaluated SCs and their physico-chemical properties(log P)(see Fig.1),C18silica stationary phases were considered for separation.Accordingly,two columns were tested:a C18Cogent bidentate silica C(4.6 mm×250 mm,4 μm)and an XTerra MS C18(4.6 mm × 250 mm,5 μm).Different mobile phases,containing mixtures of water:methanol or water:-acetonitrile,ranging between 60% and 100%(V/V)of organic solvent,as well as different column temperatures(25,30,37,40,45,50,55°C),were assessed for the HPLC separation of compounds under investigation.
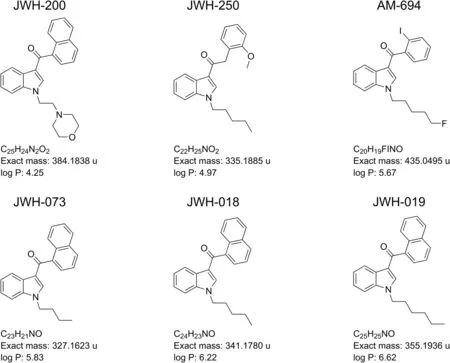
Fig.1.Structures,exact masses and log P of the synthetic cannabinoids selected for this study.
For chromatographic applications,the C18Cogent bidentate silica C is known to be advantageous because it has a limited number of non-polar silicon hydride(Si-H)groups and is effective for the separation of both hydrophobic and polar analytes[28].The best results,in terms of chromatographic efficiency and baseline resolution,were obtained by elution with acetonitrile:water(75:25,V/V)at 37°C;these conditions allowed the complete separation of AM-694,JWH-250,JWH-073,JWH-018,JWH-019,and JWH-200 in approximately 14 min.Conversely,on the XTerra MS C18,a conventional silica phase end-capped column,analyte separation was accomplished within 8 min,but two analytes(JWH-200/AM-694 and JWH-250/JWH-073)were not separated at the baseline.Figs.2A and B present the separation of SCs on the bidentate and end-capped C18stationary phases,respectively.
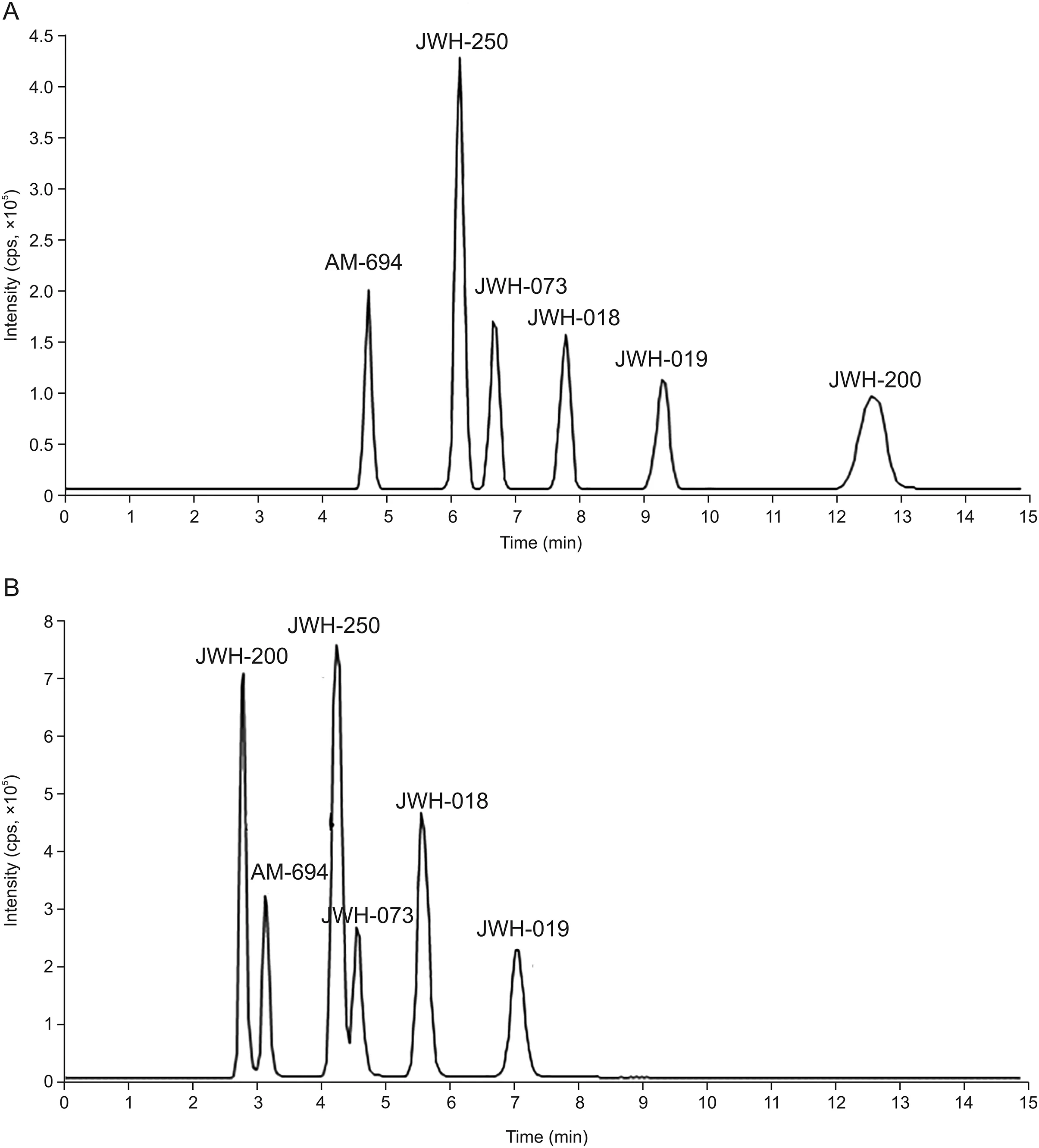
Fig.2.Chromatographic separation of the selected cannabinoids on(A)a bidentate C18silica hydride phase and on(B)an end-capped C18stationary phase.
As demonstrated,the selectivities of the two columns differed,especially for JWH-200,the analyte that possesses the lowest log P value.On the end-capped column,the interaction took place according to the compound hydrophobicity(JWH-200 was the first to elute),while contrasting behavior was observed on the bidentate column,from which JWH-200 was the last SC to elute.The different selectivity achieved for this compound with the bidentate stationary phase can be explained by considering the basicity of the morpholine nitrogen(the other SCs are neutral compounds;see Fig.1)and other factors such as steric hindrance and/or bonding of octadecyl to silica.Furthermore,it is interesting to highlight that the chromatographic retention of JWH-200 on the Cogent column was particularly influenced by temperature.In general,it is well known that an increase in column temperature can influence analyte retention,mainly due to changes in the mobile phase viscosity[29].As expected,in this study,a temperature increase in the range of 25-55°C decreased retention times of all evaluated compounds on both C18columns.However,the effect was more pronounced for JWH-200 on the bidentate column:it was the most retained analyte for temperatures less than 45°C;however,above this value,it showed retention similar to that of JWH-019(45°C)and JWH-018(55°C),resulting in the loss of the baseline separation.
3.2.Optimization of the extraction conditions
Oral fluid is a biological solution containing water(more than 97%),cells,enzymes,glycoproteins such as mucins,minerals,and other organic compounds(e.g.,urea,uric acid,cholesterol,vitamins,and phospholipids)[30].In addition to this complexity,oral fluid poses additional pre-analytical challenges owing to its viscosity and surface tension.Moreover,the DLLME procedures that have been applied to this matrix[25-27]have failed to employ a preliminary deproteinization step,hindering the achievement of good phase separation,as well as the recovery of the settled phase.As typically observed in DLLME,these procedures are based on the direct injection of a balanced solution composed of dispersing solvent and extraction solvent into the aqueous biological sample,resulting in the unavoidable formation of a protein precipitate[25,26].
Hence,the first experiments performed in this study were aimed at optimizing protein precipitation by testing different analytical solutions.In all tests,0.5 mL aliquots of a pool of oral fluid were used and spiked with the target analytes at 2 ng/mL.Chloroform(100μL)and other organic solvents(acetonitrile,THF,ethanol,or ethyl acetate)were used as extraction and dispersing solvents and vortexed for 5 min to form the cloudy solution.
Initially,the oral fluid was directly centrifuged at 12,500 g for 5 min.This approach allowed only partial protein precipitation;furthermore,when DLLME was performed,additional protein precipitate was formed following the contact of the oral fluid with the balanced solution of the dispersing(1 mL of acetonitrile)and extracting solvents.
The second approach evaluated the addition of an organic solvent(acetonitrile,THF,acetonitrile,ethanol,or ethyl acetate)to be used as the dispersing solvent in the DLLME experiments.The first attempts involved diluting the oral fluid(0.5 mL)with Milli-Q water to obtain a final volume of 5 mL.Subsequently,a solution of dispersing solvent(0.5 mL or 1 mL)and chloroform(100μL)was prepared and quickly injected into the aqueous sample.After vortexing and centrifugation,in all cases,an abundant precipitate prevented the recovery of the settled chlorinated phase at the bottom of the centrifuge tube.To avoid this inconvenience,protein precipitation was achieved by treating the oral fluid directly with the dispersing solvent.Except for ethyl acetate and partly THF(effective using only 1 mL),all other solvents(acetonitrile and ethanol)were able to precipitate proteins efficiently using either 0.5 mL or 1 mL.After centrifugation at 12,500 g for 5 min,the supernatant(1 mL or 1.5 mL,depending on the volume used)was diluted with Milli-Q water up to 5 mL.On injecting 100μL of chloroform into the diluted sample,the best extraction yields were achieved using 1 mL of acetonitrile.To further improve recoveries,both pH(4 and 9)and ionic strength(NaCl concentrations of 50,100,and 200 mg/mL)of the sample were varied;however,as expected,the results demonstrated that the effect of these parameters on obtained yields was not significant owing to the neutral nature of the SCs(pH)and the proper selection of the extraction solvent(no further advantages from the salting-out effect due to the increased ionic strength).Finally,comparative experiments(vortex and ultrasound tested for 1,2,5,and 10 min)showed that a finely dispersed solution was easily obtained after vortexing for 2 min.
During this phase of experimentation,a further attempt to increase the extraction efficiency was undertaken by testing an LTTM as the extraction solvent.Maintaining all conditions of the optimized procedure with chloroform,the effect of chlorine chloride(sesamol)3(100μL)was assessed.Unfortunately,acetonitrile was unable to disperse the LTTM in the aqueous sample,and hence,a new optimization study identified THF(1 mL)as a more suitable solvent.In this case,the pH and ionic strength were not relevant,while the best dispersion conditions were obtained by applying 5 min of ultrasound.
Fig.3 shows the comparison between the two developed DLLME methods.On employing LTTM as an extractant,recoveries ranged from 41% to 108%.Superior results were obtained with chloroform,which permitted recoveries greater than 85%.
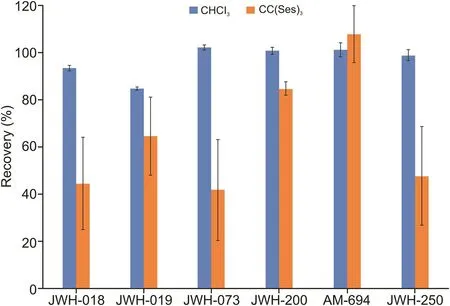
Fig.3.Comparison between two DLLME procedures based on the use of chloroform and the deep eutectic solvent chlorine chloride(sesamol)3as extractants.In this case,the chlorinated solvent offers better recovery yields for most analytes.DLLME:dispersive liquid-liquid microextraction.
3.3.Method validation
All figures of merit of the validated method are reported in Table 2.
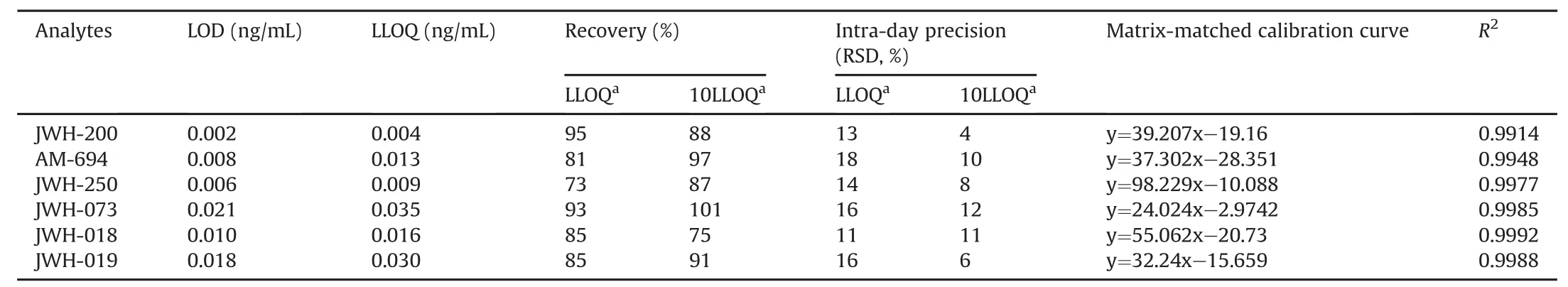
Table 2 Validation results(n=5).
Matrix-matched calibration curves were constructed by spiking five aliquots of oral fluid(each 0.5 mL)with increasing concentrations of the analytes(0.03,1,10,20,30,40,and 50 ng/mL);an unspiked aliquot was used as a “zero concentration”sample.Extraction and analysis were performed according to the procedure described in the Experimental Section.The peak area and concentration were linearly related to determination coefficients(R2)greater than 0.9914,as shown in fig.S1.
LODs and LLOQs were in the range of 2-21 pg/mL and 4-35 pg/mL,respectively,and were calculated via the analysis of five replicates,after preliminary experiments aimed at determining the spike levels detectable and quantifiable with a signal-to-noise ratio of 3 and 5,respectively.
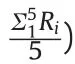
The same experiments planned for the recovery were used to determine method precision:intra-day when performed within the same analytical session,and the inter-day when executed in two different analytical sessions(over two weeks).In all cases,the relative standard deviation(RSD)was less than 18%.
3.4.Comparison with other extraction methods
The main figures of merit(recovery,precision,LOD)of the validated method were compared with those of previous methods,based on other extraction techniques and developed to isolate some SCs from oral fluid[17-21](Table 3).Overall,our procedure is appealing owing to its good general performance.The LODs were up to 10 times lower[17-21],except for a few cases for which analogous values were determined.The recoveries were higher on average,but the SPE-based method showed better yields even if JWH-018 was recovered with an anomalously high value[20].As far as precision is concerned,only two methods exhibit better figures[19,20].Nevertheless,the present method has proved to be effective at low concentrations as recovery and precision were evaluated at significantly lower spike levels(from 100 to 1000 times lower than those of the other methods[17-21]).Regarding extraction time,our procedure is more rapid and simpler.The only exceptions are the “dilute and shoot”approach[19]and the one based on“deproteinization with ice acetonitrile”[21],which,however,provided a worse clean-up of the extracts.
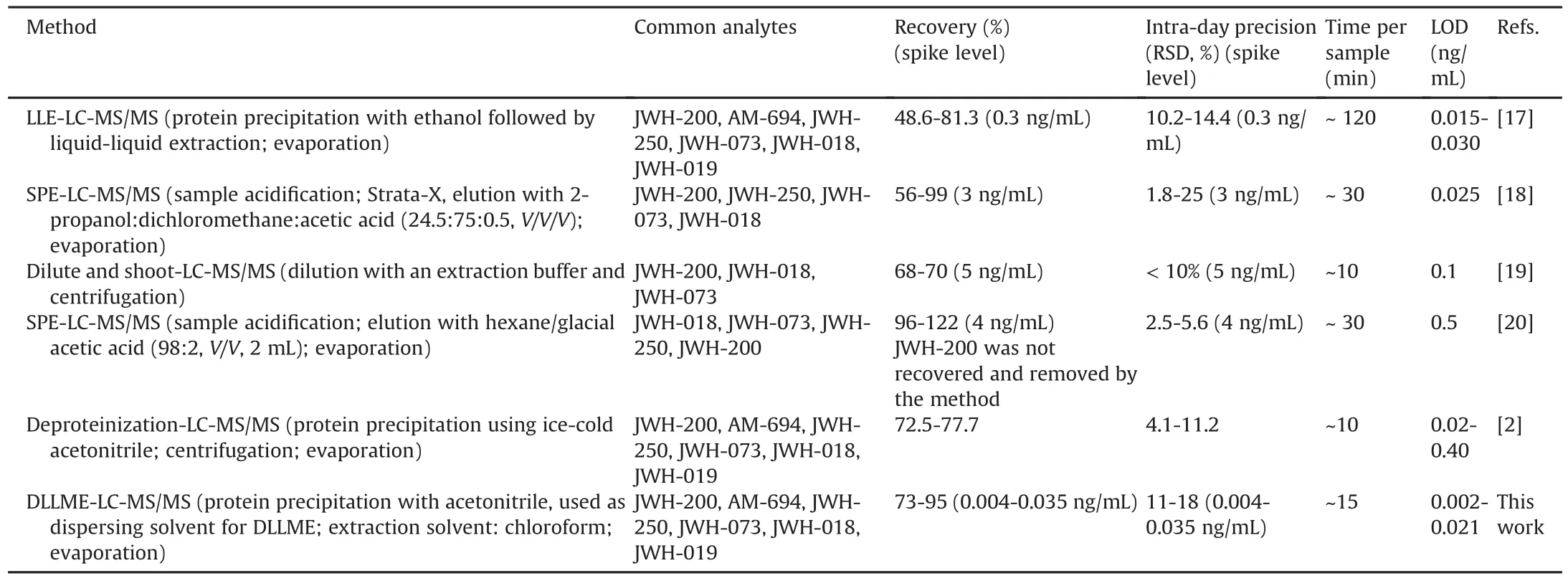
Table 3 Comparison of some recent methods through their main figures of merit.
Finally,we also performed an in-lab comparison using two different SPE-clean up procedures and the same HPLC-MS/MS instrumental method.The comparison was limited to the evaluation of recovery efficiency.The preliminary step to precipitate proteins was the same as that optimized for the DLLME procedure:0.5 mL of a spiked oral fluid(at LLOQ)was treated with 1 mL of acetonitrile and,after centrifugation,the supernatant was removed and diluted with Milli-Q water up to 50 mL.Then,the diluted supernatant was loaded onto an Oasis HLB cartridge,and the analytes were eluted with 9 mL of methanol:dichloromethane(50:50,V/V).Evaporation and reconstitution were performed as described in the DLLME procedure.The results,evaluated using three replicates,showed recoveries between 63% and 68%,except for JWH-019(39%).Another sorbent which was tested was buckypaper,used as a membrane to perform stir-disc SPE[31-33].In this case,the buckypaper disc was submerged into the diluted supernatant(50 mL)and left overnight under magnetic stirring,to favor analyte sorption;analyte desorption was performed using 9 mL of methanol:dichloromethane(50:50,V/V).Herein,the average recoveries were markedly high,ranging between 65.2% and 94.6%(three replicates).Surprisingly,JWH-200 was not recovered,probably due to its basic nature and the steric hindrance of the morpholine ring.In conclusion,the DLLME method is an advantageous alternative to perform routine analytical tasks in clinical and forensic toxicology laboratories, as demonstrated when compared with several different extraction techniques and procedures.
4.Conclusions
In the present study,an original DLLME procedure,resulting from a combination with a preliminary deproteinization step,was developed to isolate SCs from the oral fluid.The analysis of this matrix appears simple only at the surface,as the presence of peptides and proteins makes it substantially challenging.Their tendency to precipitate hinders the DLLME process,while their occurrence in the final extract can stress the HPLC-MS system,with a negative impact on both the column life and the instrumental signal.Hence,their exhaustive removal was performed in the developed procedure.To this end,acetonitrile was identified as a suitable solvent to perform the dual role of protein-denaturing and dispersing agents.High recovery efficiencies were obtained by selecting chloroform as the extraction solvent,which was more efficient than the LTTM chlorine chloride(sesamol)3.On comparing SPE using Oasis HLB and buckypaper,the DLLME procedure was proved to be advantageous owing to its general performance and rapidity.Finally,this study highlights the superior selectivity of the bidentate C18silica hydride column and the particular chromatographic behavior of JWH-200,which distinguishes itself from other SCs as less hydrophobic and slightly basic.The isocratic separation on this column was set up to separate all analytes at the baseline within 14 min,providing a method that,as a whole,is advantageous for clinical and forensic routine analyses.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the Sapienza University of Rome through the project RICERCA 2019 (protocol number:RG11916B6451D44A).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.11.004.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Molecular detection of SARS-CoV-2 being challenged by virus variation and asymptomatic infection
- The potential of miRNA-based therapeutics in severe acute respiratory syndrome coronavirus 2(SARS-CoV-2)infection:A review
- Potential treatment with Chinese and Western medicine targeting NSP14 of SARS-CoV-2
- Accurate and sensitive determination of hydroxychloroquine sulfate used on COVID-19 patients in human urine,serum and saliva samples by GC-MS
- Fast saccharide mapping method for quality consistency evaluation of commercial xylooligosaccharides collected in China
- Identification and quantification of the bioactive components in Osmanthus fragrans roots by HPLC-MS/MS