利用无溶剂研磨反应制备3-取代异吲哚啉-1-酮类化合物
2021-07-14王平安姚琳张东旭聂慧芳李穆琼姜茹
王平安,姚琳,张东旭,聂慧芳,李穆琼,姜茹
空军军医大学药学系,药物化学与药物分析学教研室,西安 710032
3-取代异吲哚啉-1-酮是许多药物和具有生理活性的天然产物的重要结构单元(图1)[1],含有该结构单元的药物如帕戈隆(Pagoclone)、帕嗪克隆(Pazinaclone)等已经用于治疗如口吃、焦虑、失眠等一些精神性疾病。经爱思唯尔(Elsevier)公司化学数据库Reaxys®查询可知,含有这类骨架的化合物达29000多个,其中有生理活性的化合物占到80%以上,表现出良好的抗炎、抗癌、抗真菌、抗病毒、抗精神疾病等药理活性,如多巴胺D4受体激动剂(S)-PD-172938[2]和苯二氮卓类受体拮抗剂(R)-JM 1232[3]等。3-取代异吲哚啉-1-酮结构单元还存在于许多具有生理活性的天然分子中,如生物碱Lennoxamine、Chilenine、Taliscanine等。这些分子被证实具有抗癌、抗氧化等作用。3-取代异吲哚啉-1-酮骨架的合成方法分为金属催化和有机催化两类[4],这些方法绝大多数是在有机溶剂中进行的,反应时间较长(2–72 h),有些还使用了对水、空气敏感的试剂,所得产物绝大多数需要柱层析分离纯化,操作繁琐,难以规模化合成。
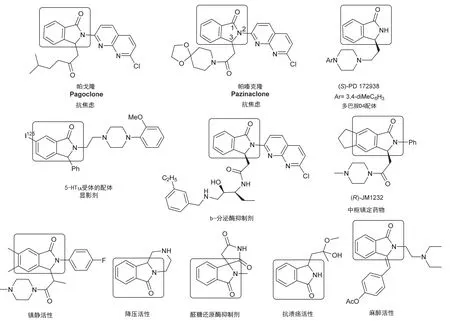
图1 含有3-取代异吲哚啉-1-酮结构单元的药物及生物活性分子
绿色合成化学[5]的发展涌现出一大批优异的反应模式,光照、超声、微波、加压、研磨等物理手段介入有机合成化学,为各类化合物的绿色制备提供便捷。研磨反应(Grinding Reaction)是机械化学(Mechanochemistry)[6]的分支,分为两种方法,一种是将反应物在研钵里混匀并研磨;另一种是将反应物加入到球磨机里,使用不同直径的研磨球进行球磨。上述两种方法绝大多数无需使用易挥发的有机溶剂,操作简单、反应时间短且产率高,在有机物合成、纳米材料制备以及药物辅料生产中应用广泛。
研磨反应在有机合成中的应用非常广泛。然而,在目前大学化学的实验教材中,均未见到使用研磨反应制备有机化合物的实例。近期我们发展了利用简单研磨快速进行Michael加成反应的方法[7]。为展示研磨反应简便高效的特点,我们设计了以2-氰基芳醛和活泼亚甲基化合物(如丙二酸二甲酯、乙酰乙酸乙酯等)为原料,在无水碳酸钾催化下,通过室温研磨反应,一步制备3-取代异吲哚啉-1-酮类化合物的实验,期望能够抛砖引玉,将研磨反应带入到本科生化学实验课堂。旨在让学生建立绿色合成化学理念,开拓知识视野,熟练运用薄层色谱(TLC)监测反应进程和产物纯度,了解有机合成化学的发展前沿,激发学习兴趣。
1 实验目的
(1) 熟悉相关文献检索网站(如美国化学会ACS,英国皇家化学会RSC等),学习中英文相关文献检索方法,通过查阅文献了解含有3-取代异吲哚啉-1-酮骨架化合物的生物及药理活性;
(2) 了解绿色有机合成化学的发展现状,加深对研磨法制备有机化合物的认识;
(3) 熟悉Aldol串联环化反应机理,掌握3-取代异吲哚啉-1-酮类化合物的制备方法;
(4) 掌握薄层色谱监测反应的实验操作;
(5) 理解核磁共振仪的基本操作和相应的谱图分析。
2 实验原理
利用2-氰基芳甲醛和活泼亚甲基化合物构建3-取代异吲哚啉-1-酮的反应机理比较复杂[8],如图2所示。第一步,活泼亚甲基在催化剂作用下与2-氰基芳甲醛发生Aldol反应,生成加成中间体A;第二步,加成中间体A的羟基分子内亲核进攻邻位―CN,生成环亚胺B,紧接着发生重排(rearrangement),形成开环中间体C;第三步,C发生分子内Michael加成,得到3-取代异吲哚啉-1-酮骨架。这几步紧密相连,互为基础,共同完成环化反应。
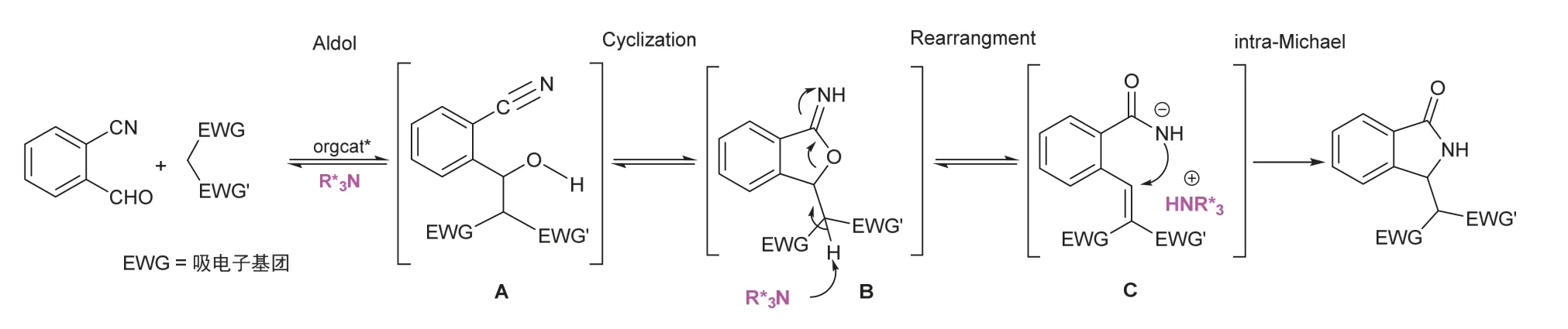
图2 Aldol串联环化反应机理
3 仪器和试剂
仪器:核磁共振仪(Bruker AV 400 Spectrometer,CDCl3为溶剂,TMS为内标);高分辨质谱仪(Bruker microTOF-Q II Mass Spectrometer,ESI-HRMS);高效液相色谱仪(HPLC,安捷伦1260);熔点仪(JH30)全自动熔点仪。
试剂:2-氰基苯甲醛(分析纯),丙二酸二甲酯(分析纯),丙二酸二乙酯(分析纯),丙二酸二异丙酯(分析纯),丙二酸二苄酯(分析纯),乙酰丙酮(分析纯),乙酰乙酸乙酯(分析纯),二氯甲烷(分析纯),石油醚(精馏级),乙酸乙酯(精馏级),三乙胺(Et3N,分析纯),二异丙基乙基胺(iPr2NEt,分析纯),三乙烯二胺(DABOC,分析纯),1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU,分析纯),三苯基膦(PPh3,分析纯),氢氧化钠(NaOH,分析纯),碳酸钠(Na2CO3,分析纯),无水碳酸钾(K2CO3,分析纯),薄层色谱硅胶板(F254)和柱层析硅胶(200–300目)均购于青岛海洋化工有限公司。
4 实验内容
4.1 筛选实验
我们选用2-氰基苯甲醛和丙二酸二甲酯为模板底物,使用三乙胺(Et3N)、二异丙基乙基胺(iPr2NEt)、三乙烯二胺(DABOC)、1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)、三苯基膦(PPh3)、氢氧化钠(NaOH)、碳酸钠(Na2CO3)、碳酸钾(K2CO3)等8种有机和无机碱,对无溶剂研磨反应制备3-取代异吲哚啉-1-酮的可能性进行了验证,实验结果如表1所示。
实验操作如下:
称取1.0 mmol 2-氰基苯甲醛(132 mg)、1.1 mmol丙二酸二甲酯(135 mg)和表1相应数量的碱,置于6 cm口径天然玛瑙研钵中,混合均匀,室温研磨相应时间。用薄层色谱监测反应情况。待反应完全后,用15 mL二氯甲烷转移反应混合物。对使用固体无机碱或盐的反应液过滤,蒸干溶剂得产品;对使用液体有机碱的反应液先用1 mol·L−1HCl 5 mL洗涤一次,再分别用水(5 mL)和饱和食盐水(5 mL)洗涤一次,无水硫酸钠干燥,蒸干溶剂后得产品,分别计算产率。
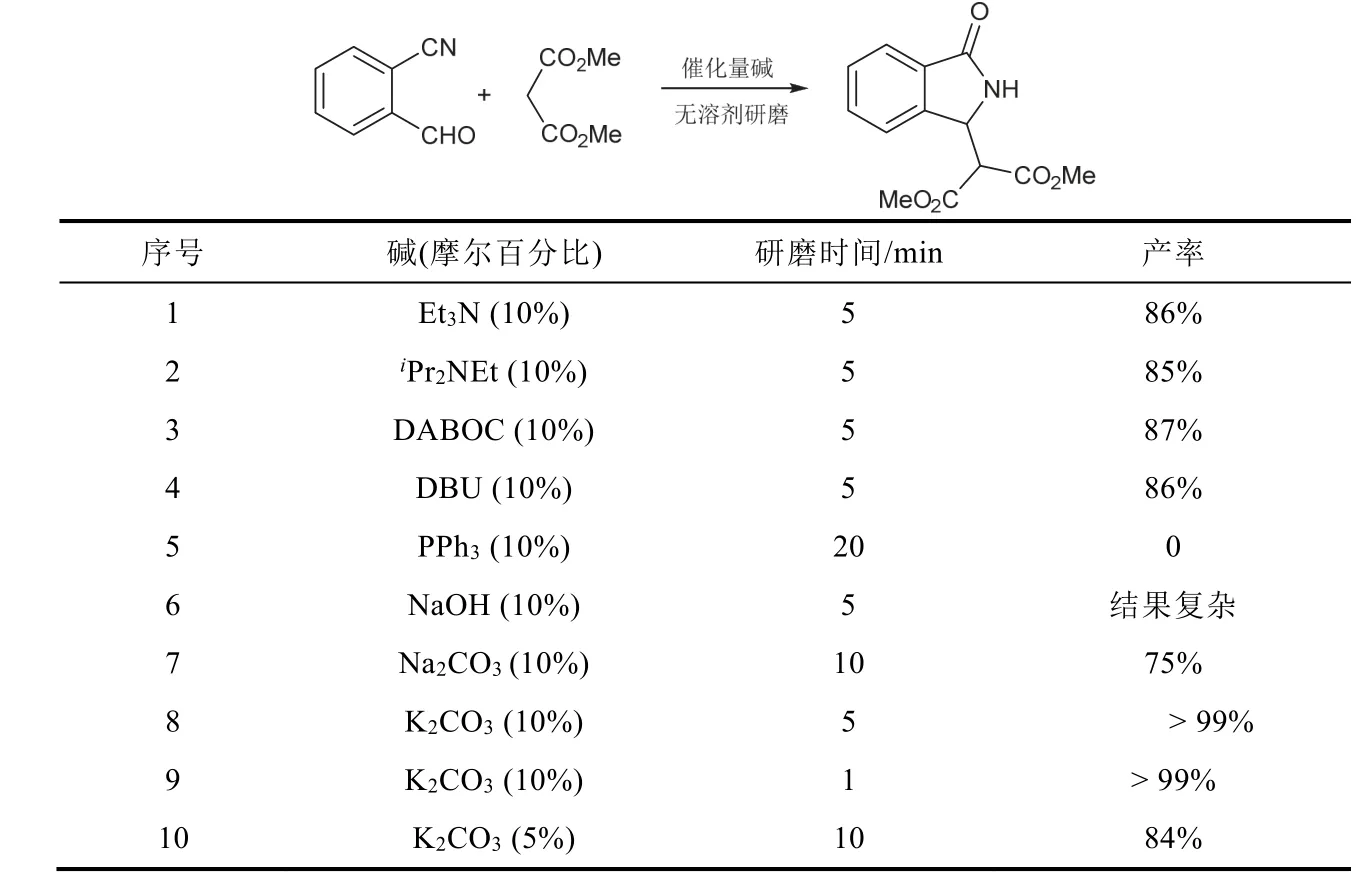
表1 筛选实验结果
表1的筛选实验结果表明,除PPh3以外,其他有机碱都可以催化模板反应,且能获得较高产率,但由于所使用的有机碱均为液体,不方便准确量取,因此选用固体NaOH作为催化剂,相同条件下导致反应结果复杂,无法得到相应产物;将催化剂换成Na2CO3时,可获得75%的产率;当使用10% (x,摩尔百分比)的K2CO3催化该反应时,室温研磨1 min即可获得大于99%的产率,且无机盐一般难溶于有机溶剂,很容易从反应体系中除去,所以最终选用K2CO3作为最优催化剂。
4.2 主体实验
称取1.0 mmol 2-氰基芳甲醛、1.05 mmol活泼亚甲基化合物和14.0 mg无水K2CO3(10% (x)),置于6 cm口径天然玛瑙研钵中,混合均匀,室温研磨1–10 min。薄层色谱监测显示反应完全(展开剂石油醚/乙酸乙酯,体积比为2 : 1,产物的Rf值为0.1–0.3之间)。用10 mL二氯甲烷分批转移干净反应混合物,过滤,浓缩,无需柱层析即可获得产品。通过上述无溶剂研磨反应,一共获得了15个产物(图3),显示了该方法的稳定性、普适性和便捷性。所得产物用核磁共振分析确定其结构。
随着综合含水的上升,每采出1 吨油所需要的产液量大幅度增加,用于补充地层能量的注水量也相应增加,导致举升成本、处理成本、注入成本增加,单位生产成本上升。
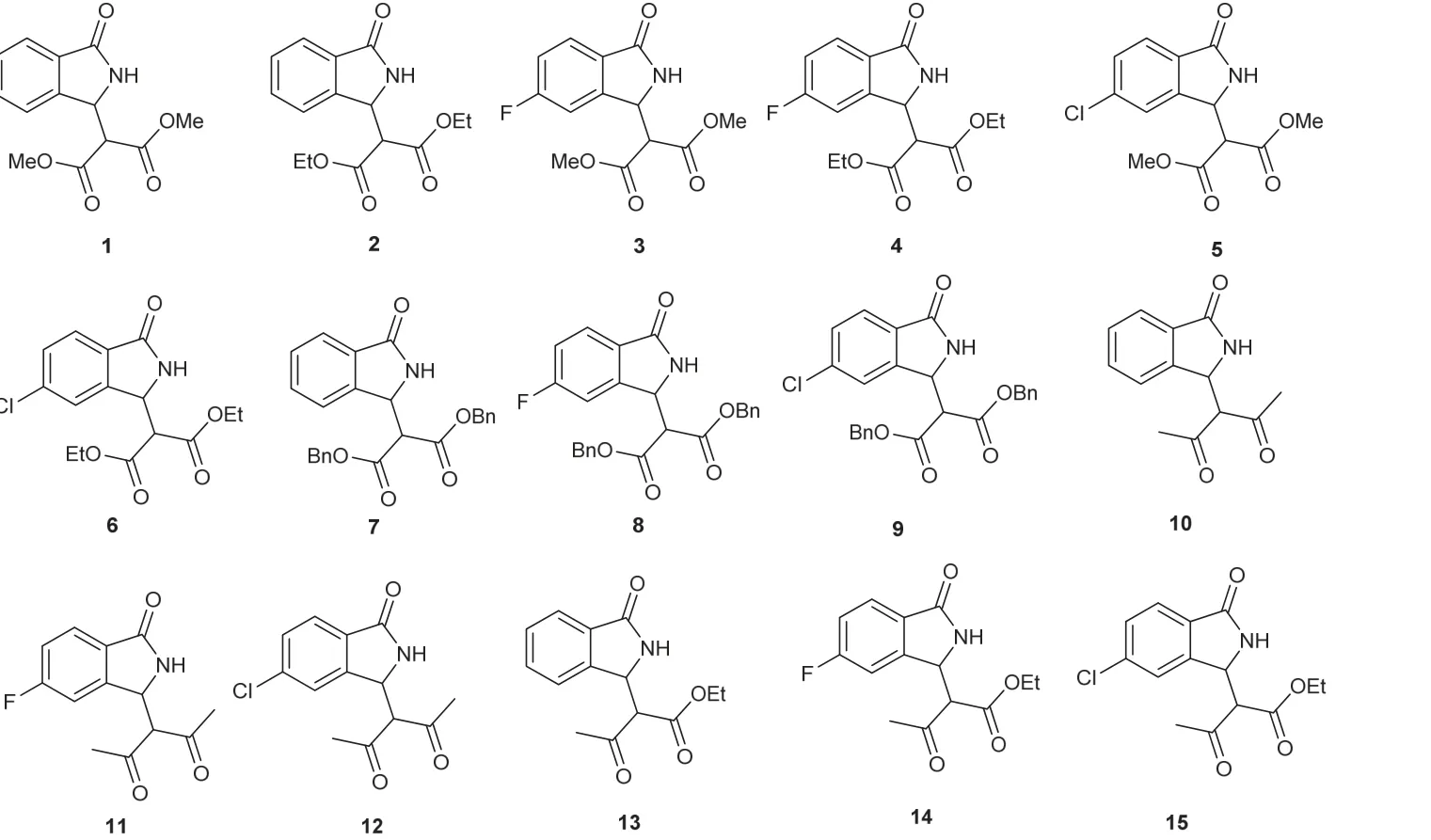
图3 无溶剂研磨反应制备3-取代异吲哚啉-1-酮类化合物结构
4.3 实验结果
我们分别以“2-氰基苯甲醛+丙二酸二甲酯”“2-氰基苯甲醛+丙二酸二乙酯”“4-氟-2-氰基苯甲醛+丙二酸二乙酯”和“2-氰基苯甲醛+乙酰乙酸乙酯”为例,进行了无溶剂研磨放大实验(5–10 mmol规模,图4),它们的具体实验结果如下。

图4 无溶剂研磨反应放大操作
4.3.1 反应实例1:2-氰基苯甲醛+丙二酸二甲酯(式-1)
称取140 mg无水K2CO3(10 mol%),置于12 cm口径的白色陶瓷研钵中,快速研磨成细粉末(约10 s),迅速加入事先称取好的10.0 mmol 2-氰基苯甲醛和10.5 mmol丙二酸二甲酯(式-1)。室温继续研磨,反应混合物变得粘稠,1 min后液体原料完全消失,此时反应混合物结块(注意:反应轻微放热);再研磨2 min至粉末状。薄层色谱监测显示反应完全(展开剂石油醚/乙酸乙酯,体积比为2 : 1,产物1的Rf值为0.20,图5)。先将固体粗产物粉末转移出研钵(粗产物的粉红色来自原料2-氰基苯甲醛),再用20 mL二氯甲烷分批转移干净反应混合物。将粗产物粉末用40 mL二氯甲烷溶解,合并后通过1.0 cm厚的200–300目硅胶抽滤,用10 mL二氯甲烷洗涤硅胶层,得浅黄色至黄色清亮滤液。浓缩滤液,即可得到产物。所得产物用核磁共振氢谱确定其结构(图6)。淡黄色至黄色固体,2.62 g,产率> 99%。

图5 产物1的TLC分析
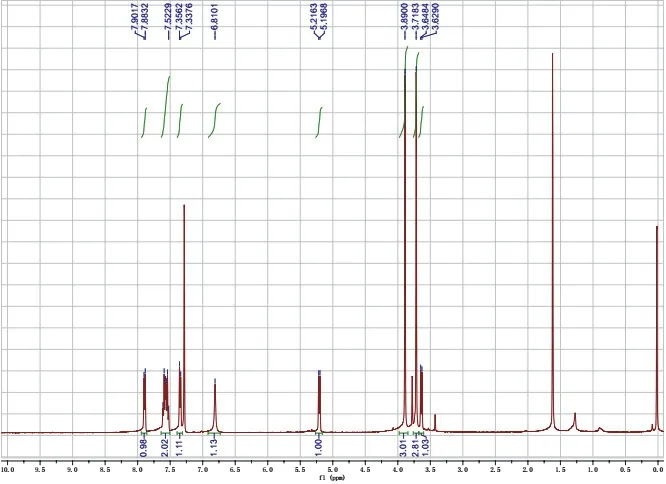
图6 产物1的1H NMR图谱
1H NMR (400 MHz, Chloroform-d)δ7.89 (d,J= 7.4 Hz, 1H),7.61–7.52 (m, 2H),7.35 (d,J= 7.4 Hz,1H),6.81 (s, 1H),5.21 (d,J= 7.8 Hz, 1H),3.89 (s, 3H),3.72 (s, 3H),3.64 (d,J= 7.8 Hz, 1H)。
4.3.2 反应实例2:2-氰基苯甲醛+丙二酸二乙酯(式-2)
操作步骤(式-2)同反应实例1。反应较丙二酸二甲酯慢,需研磨7–10 min,Rf= 0.2 (展开剂石油醚/乙酸乙酯,体积比为2 : 1,图7)。所得产物用核磁共振氢谱确定其结构(图8)。淡黄色至黄色固体,2.88 g,产率> 99%。1H NMR (400 MHz, Chloroform-d)δ7.89(d,J= 7.4 Hz, 1H),7.61–7.52(m, 2H),7.39 (d,J= 7.36 Hz, 1H),6.81(s, 1H),5.19 (d,J= 7.16 Hz, 1H),4.39–4.31 (m, 2H),4.14 (q,J= 7.12 Hz,2H),3.65 (d,J= 7.28 Hz, 1H),1.35 (t,J= 7.12 Hz, 3H),1.14 (t,J= 7.12 Hz, 3H).

图7 产物2的TLC分析
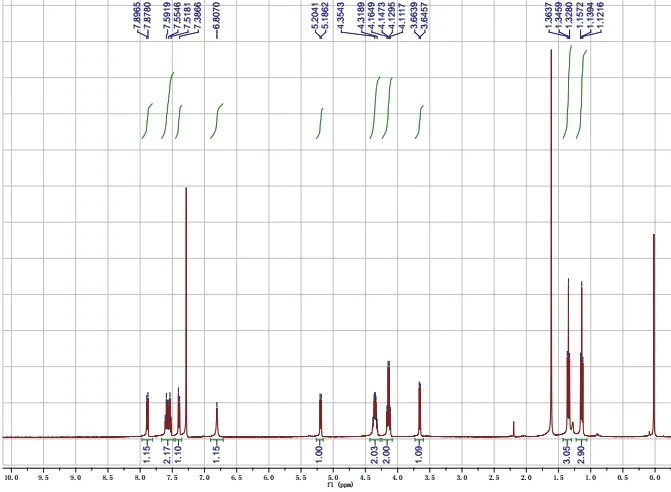
图8 产物2的1H NMR图谱
4.3.3 反应实例3:4-氟-2-氰基苯甲醛+丙二酸二甲酯(式-3)
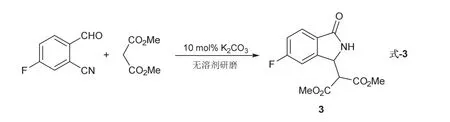

图9 产物3的TLC分析

图10 产物3的1H NMR图谱
4.3.4 反应实例4:2-氰基苯甲醛+乙酰乙酸乙酯(式-4)
操作步骤(式-4)同反应实例1 (5 mmol规模)。反应较丙二酸二甲酯慢,需研磨7–10 min,Rf= 0.2(展开剂石油醚/乙酸乙酯,体积比为2 : 1,图11)。所得产物为非对映异构体混合物(4/1)。核磁共振氢谱确定其结构(图12)。淡黄色至黄色低熔点固体,1.30 g,产率> 99%。1H NMR (400 MHz, Chloroformd)δ7.88, 7.74 (d,J= 6.6 Hz, 1H), 7.59–7.50 (m, 2H), 7.32 (d,J= 8.2 Hz, 1H), 6.68, 6.21 (s, 1H), 5.26 (d,J= 7.6 Hz, 1H), 4.35, 4.15 (q,J= 7.2 Hz, 2H ), 3.82, 3.86 (d,J= 9.6 Hz, 1H), 2.39, 2.22 (s, 3H), 1.16, 0.98(t,J= 7.2 Hz, 3H)。

图11 产物13的TLC分析
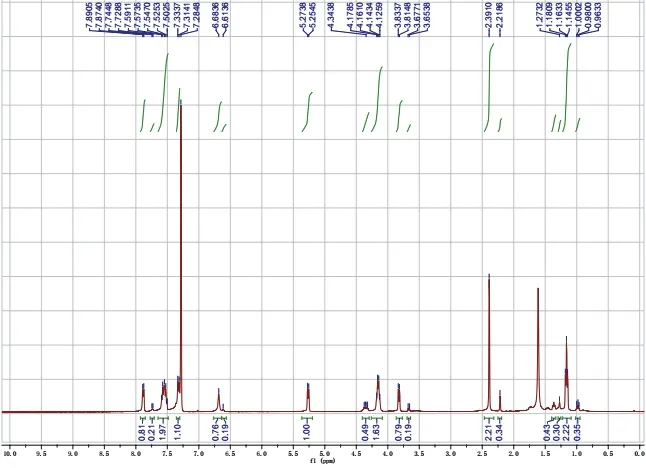
图12 产物13的1H NMR图谱
5 讨论
5.1 底物对反应的影响
活泼亚甲基化合物种类繁多,对丙二酸酯类化合物而言,甲酯的反应速率大于乙酯,乙酯的反应速率大于异丙酯,叔丁酯则几乎不发生上述研磨反应;对乙酰乙酸乙酯和乙酰丙酮而言,前者的反应速率大于后者;硝基乙酸乙酯、丙二腈、氰乙酸甲酯等活泼亚甲基化合物也能发生上述研磨反应。亚甲基两侧基团相同时(对称亚甲基化合物,如丙二酸二甲酯),可得到一对外消旋体;当亚甲基两侧基团不同时(非对称亚甲基化合物,如乙酰乙酸乙酯),会得到4种异构体,但通常以一对外消旋体为主要产物。
5.2 无水K2CO3用量对反应的影响
无水K2CO3的用量控制在10%–15% (x)之间为宜,减少用量会导致反应速率变慢;增加用量则会导致产物收率降低,可能是因为酯基水解的缘故。
6 实验注意事项
(1) 无水K2CO3在空气中易吸水,要快称快用。
(2) 2-氰基芳醛质轻,粉末容易飞溅,易引起呼吸道不适(如咳嗽等),应在通风橱中称量和使用。
(3) 建议反应规模控制在5 mmol以下,以免造成污染和浪费。
7 结语
利用无溶剂研磨反应制备3-取代异吲哚啉-1-酮类化合物,相比较传统的使用溶剂搅拌反应而言,要方便快捷。使用的活泼亚甲基化合物价廉易得,既可大量操作,也能微量反应。所得产物可以进一步水解脱羧[9]或进行其他官能团转化,非常适合绿色有机合成实验教学。
