自由基型光引发剂简介及研究进展
2021-07-14李雪纯孙芳
李雪纯,孙芳
北京化工大学化学学院,北京 100029
在21世纪,以光作为能量来源进行反应的绿色化学受到人们瞩目。大多数光化学参与的生产过程能有效避免传统生产方法的高能耗问题,这对于实际的工业生产而言至关重要。光聚合技术,作为这一方面的代表,是一种在温和条件下光聚合体系中的光引发剂被光激发,生成承担着引发单体聚合作用的活性物种,使体系由液态快速变为固态的低成本绿色技术。光聚合体系通常由光引发剂、低聚物、单体和负责满足材料特殊使用要求的助剂构成。光引发剂直接决定着光聚合速率的快慢,同时也影响着聚合后材料的物理力学性能,因此光引发剂在整个光聚合过程及体系中扮演着关键角色。按照产生活性物种的类型,光引发剂可分为自由基型光引发剂和阳离子型光引发剂。相比于阳离子型光引发剂,自由基型光引发剂具有引发速度快、种类多及价廉易得的优势,是目前光聚合体系中应用最广泛的类型,因此,本文将从其分类、引发机理和研究进展三个方面来详细介绍自由基型光引发剂。
1 自由基型光引发剂的种类
光引发剂产生自由基的方式因光引发剂的结构不同而有所区别,按照产生自由基机理的不同,自由基型光引发剂可以分为两大类。
第一类称为裂解型光引发剂,这类光引发剂大都具有芳香烷基酮结构,比如苯偶酰及其衍生物类[1–3]、酰基膦氧化物类[4–6]、α-羟基酮及其衍生物类[7,8],如图1所示。它们在吸收光子能量之后,由基态跃迁至激发单线态,通过系间窜跃(intersystem crossing,ISC)到达激发三线态,处于激发态的分子通过化学键的均裂形成具有引发能力的活性自由基[7,9,10]。图2以双(2,4,6-三甲基苯甲酰基)二苯基氧化膦(BAPO)为例,展示了裂解型光引发剂产生自由基的机理[11]。
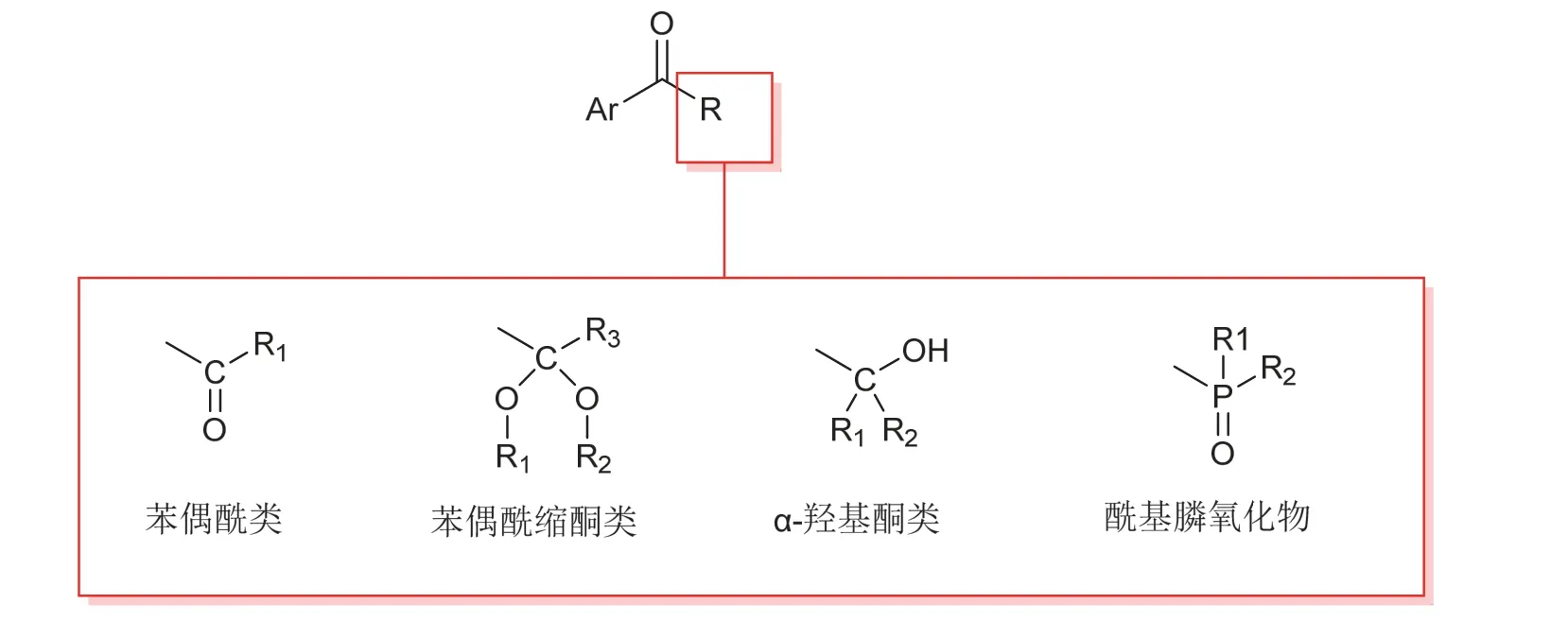
图1 部分裂解型光引发剂结构
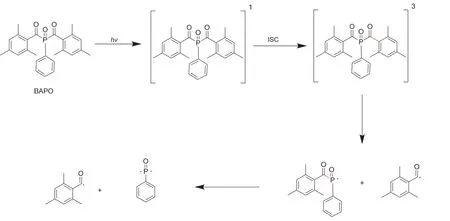
图2 BAPO光解产生自由基示意图[11]
第二类则是夺氢型光引发剂,一般情况下它们通过单分子反应无法产生活性自由基,需与另一分子助剂结合使用,构成多组分光引发体系(光引发剂/助剂)。这类光引发剂以芳香酮化合物为主[12–15],所用的助剂多为叔胺、硅烷等[10,16],图3展示了部分夺氢型光引发剂。图4以二组分光引发体系硫杂蒽酮(TX)/三乙胺(Et3N)为例来阐释生成活性自由基的机理[16,17]:在受到特定波长的光照后,TX由基态跃迁至激发态,与尚在基态的Et3N上N的孤对电子作用,使电子从胺转移到TX上,形成激基复合物(exciplex)。之后位于N的α位的质子转移,生成两种自由基,其中羰基自由基因为空间位阻和未成对电子的离域作用,丧失了对单体的引发活性,只有胺烷基自由基能引发单体聚合。
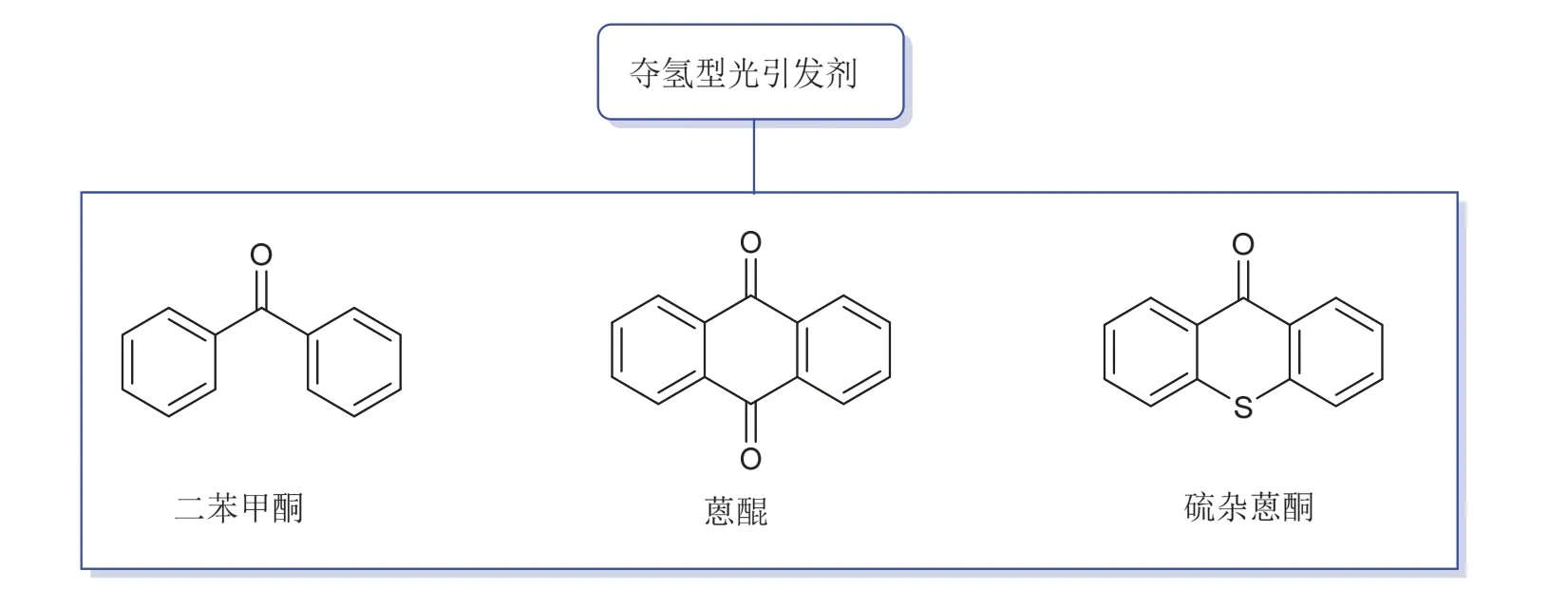
图3 部分夺氢型光引发剂结构
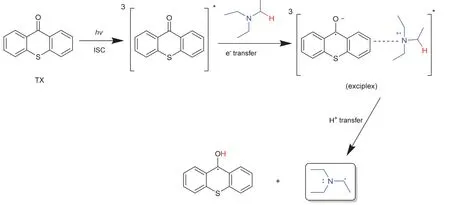
图4 TX/Et3N体系生成自由基示意图[16]
2 研究进展
现如今,光聚合技术在工业生产和学术研究上有着广泛应用,但是这项技术尚存在着诸如氧气猝灭活性物种阻碍单体聚合、残留的光引发剂及其光解产物易从聚合产品中迁移到外界环境、体系在聚合后的体积收缩等问题。特别是已有研究[18,19]发现在外界环境如电子垃圾回收设施和邻近室内粉尘中以及人体内检测到了多种光引发剂,鉴于部分光引发剂的致癌可能性,开发新一代具有低迁移能力、低毒性的光引发剂已经迫在眉睫。
在光聚合技术中应用的光源也需转变。利用紫外波段的光进行引发的聚合技术以其反应效率高的优势,在光聚合领域有着较大占比,但尚存局限性。首先,紫外线波长较短,所能穿透的深度较低难以实现深层固化,图5[20]表明光源波长越长,穿透深度越大,当光源由紫外光变为红光时,穿透深度达到厘米级别,这对于厚填充材料的固化有着重要意义;其次,紫外线会对人体的皮肤和眼睛造成伤害。针对这两点问题,将波长大于400 nm的可见光应用于光聚合领域的呼声越来越高。随着发光二极管(LED)和激光光源的飞速发展,可见光下的光聚合已经成为可能。然而,传统的光引发剂难以吸收可见光,不能在其照射下引发聚合。因此,开发适用于更长波长的可见光区域的光引发剂成为研究热点。
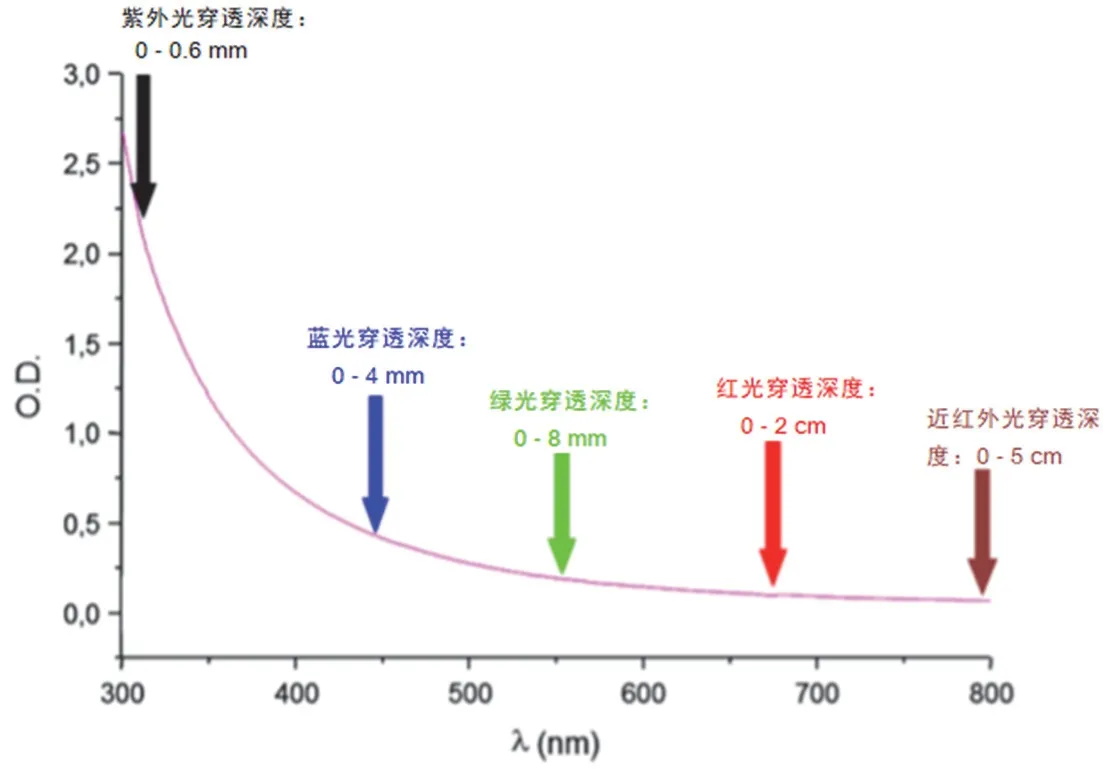
图5 聚苯乙烯乳胶(平均直径112 nm)的紫外-可见光散射和计算的选定光子穿透率[20]
以无毒性、无刺激性气味的水作为光聚合体系的稀释剂,克服了使用活性单体或有机溶剂作为稀释剂带来的挥发性化合物排放的问题。随着国家对于环境保护要求的提高,水性光聚合技术获得的关注度逐渐升高,应用领域已经扩展到生物医疗等领域[21,22]。此外,水性光聚合体系所含氧气浓度低于油性光聚合体系,这大大降低了氧气对于自由基光聚合的抑制作用[23]。相应地,针对水性光引发剂的设计与合成,成为光引发剂研究的另一个发展前沿方向。
下面将从可见光光引发剂、水性光引发剂和低迁移的大分子光引发剂三个角度来综述近阶段光引发剂的研究进展。
2.1 可见光光引发剂
提高光引发剂在可见光区域的吸收、红移其吸收带主要依靠扩大分子内的共轭结构或引入推拉电子基团来实现。这里包括了对新型染料(咔唑、萘酰亚胺等)和紫外光引发剂的结构改性。由于可见光的光子能量较低,其提供的能量难以导致化学键的均裂,因此这一方向主要集中在夺氢型光引发剂特别是多组分光引发体系的开发上,关于裂解型光引发剂的报道较少。
2.1.1 新型染料类光引发剂
咔唑是多环芳烃的一种,由一个含氮的五元杂环两侧各稠合一个苯环构成,见图6的CB。咔唑有着良好的刚性平面结构,由于五元环中N原子的参与,电子离域范围从单个苯环扩大到整个刚性平面上,具有富电子性质。这使得咔唑可与拉电子结构相连,形成“推拉”型染料,增强染料在可见光区域的吸收。Lalevée等[24]首次将这一理念应用于光引发体系的设计中:通过碳碳双键连接推电子的咔唑结构和拉电子的巴比妥酸/硫代巴比妥酸,即图6中的CB1和CB2。由于CB1和CB2都具有明显的分子内电荷转移性质,它们在350–500 nm有着良好吸收,能与457 nm、473 nm的激光二极管或462 nm的LED光源匹配。实际的自由基光聚合实验结果表明,CB1与助剂构成的多组分光引发体系无法在上述三种光源的照射下引发羟甲基丙烷三丙烯酸酯(TMPTA)的自由基光聚合,而CB2与Ph2I+、甲基二乙醇胺(MDEA)、乙烯基咔唑(NVK)等助剂构成的光引发体系则能展现出较高的引发能力。通过比较CB1和CB2的溶剂化变色能力,作者发现溶剂化变色与溶剂极性的线性相关程度与染料的光引发能力正相关。这意味着可以利用溶剂化变色能力来预测“推拉”型染料的光引发能力。之后Lalevée等[25]设计合成了四种咔唑基染料CB3–6,它们在近紫外区和可见光区有着较高的摩尔消光系数,特别是CB6,结构中的胺基和硝基的推拉电子效应令其吸收峰红移且可见光区域的吸收带变宽。CB3–6分别与助剂构成的多组分体系在LED光源(405、455、477 nm)照射下,对于环氧树脂和丙烯酸酯类单体表现出了良好的引发性能,特别是CB3–6/Ph2I+体系引发阳离子聚合的能力超越了BAPO/Ph2I+体系,这使得CB3–6/Ph2I+体系还能应用于3D打印实验。
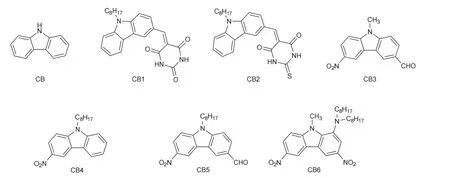
图6 咔唑及其衍生物结构
萘酰亚胺类化合物(图7)分子内具有两个苯环和一个含氮杂环稠合形成的刚性平面和较大的π电子离域范围,这类化合物还具有良好的化学稳定性、较大的斯托克斯位移和高荧光量子产率,常被设计为荧光探针,用于H2S、H2O2、HClO等物质的检测[26–28]。通过在萘环上引入不同电子性质的取代基,可以调控这类化合物的光物理、光化学性质,此外与N相连的R1基团的多样化则依靠相应的酸酐与胺反应实现,这使得萘酰亚胺逐渐成为设计光引发剂的通用平台[29]。
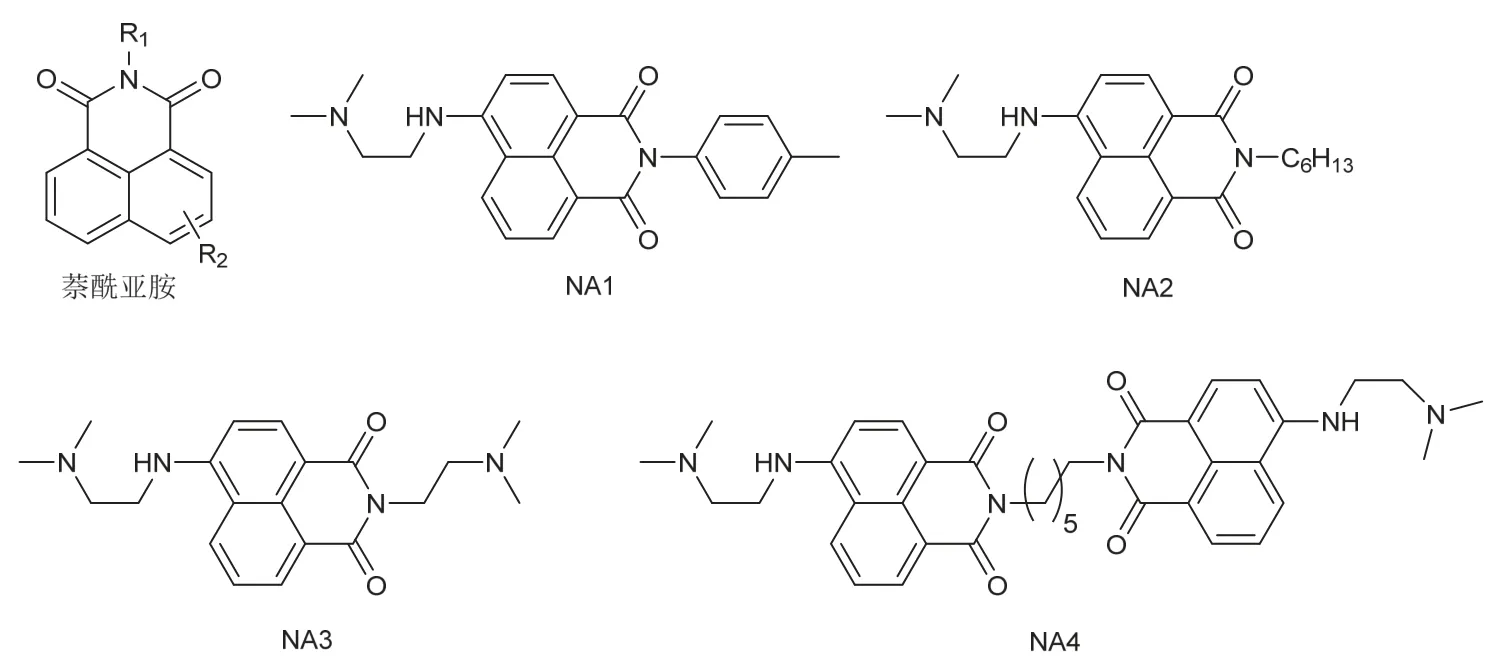
图7 1,8-萘二甲酰亚胺类化合物结构
Zhang等[30]报道了四种萘酰亚胺衍生物NA1–4,最大吸收波长均位于430 nm,由于分子中具有三级胺结构,NA1–4能作为单组分夺氢型光引发剂或与助剂相互作用,在蓝光LED照射下呈现出良好的引发性能,基于新染料的多组分光引发体系具有超越商用光引发剂的引发效率。尤其是NA1/Ph2I+/NVK体系还能用于自由基促进的阳离子聚合,以制备互穿聚合物网络。
Yang等[31]设计并合成了图8所示的萘酰亚胺衍生物NA5,通过巧妙的设计萘酰亚胺的氮原子的取代基结构,NA5不仅能作为单组分夺氢型光引发剂使用,还拥有抑制氧阻聚的功能:酯基氧的β-碳自由基被氧气捕捉后,变为反应活性较低的过氧自由基,再通过与另一分子NA5进行分子间夺氢反应或依靠较为稳定的六元环过渡态,生成新的碳中心自由基,重获引发能力。此外,由于双键的引入,NA5可以参与聚合而被锚定在聚合网络中,不易从聚合材料内部迁移到表面,减少了对外界环境的危害(毒性、气味等),因此NA5具有良好的迁移稳定性,是一种多功能的光引发剂。
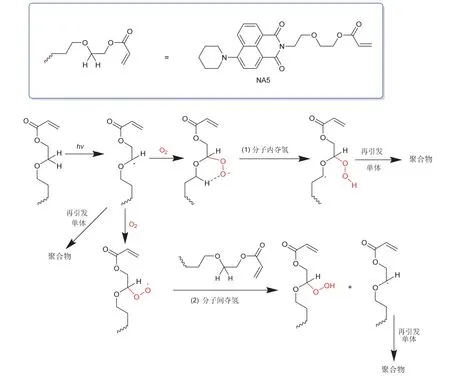
图8 推测的NA5抗氧阻聚机理[31]
本课题组在此方面也进行了研究[32]:将芳硫醚结构接在萘环的4位上,得到了一系列具有C―S弱键的萘酰亚胺衍生物NAS1–6。由于硫原子的助色作用,NASs适用于蓝光LED光源,推测的NASs的光解机理如图9所示:作为单组分裂解型光引发剂使用时,处于位置1和位置2的两种C―S键中能量更低的键发生均裂,生成相应的碳自由基和硫自由基来引发丙烯酸酯类单体的自由基聚合,其中,对于NAS1–3,两种位置C―S键能量相似,以相近的概率发生断裂产生四种自由基引发聚合;而对于NAS4–6,主要通过低能量位置1的C―S键断裂产生两种的自由基引发聚合。若无单体存在,NASs光解生成的自由基重新结合为NASs,NASs/Iod体系还能引发环氧单体的阳离子聚合。同时,NASs与单体组成的光聚合体系在日光下的稳定性好,这有利于NASs的储存和运输。
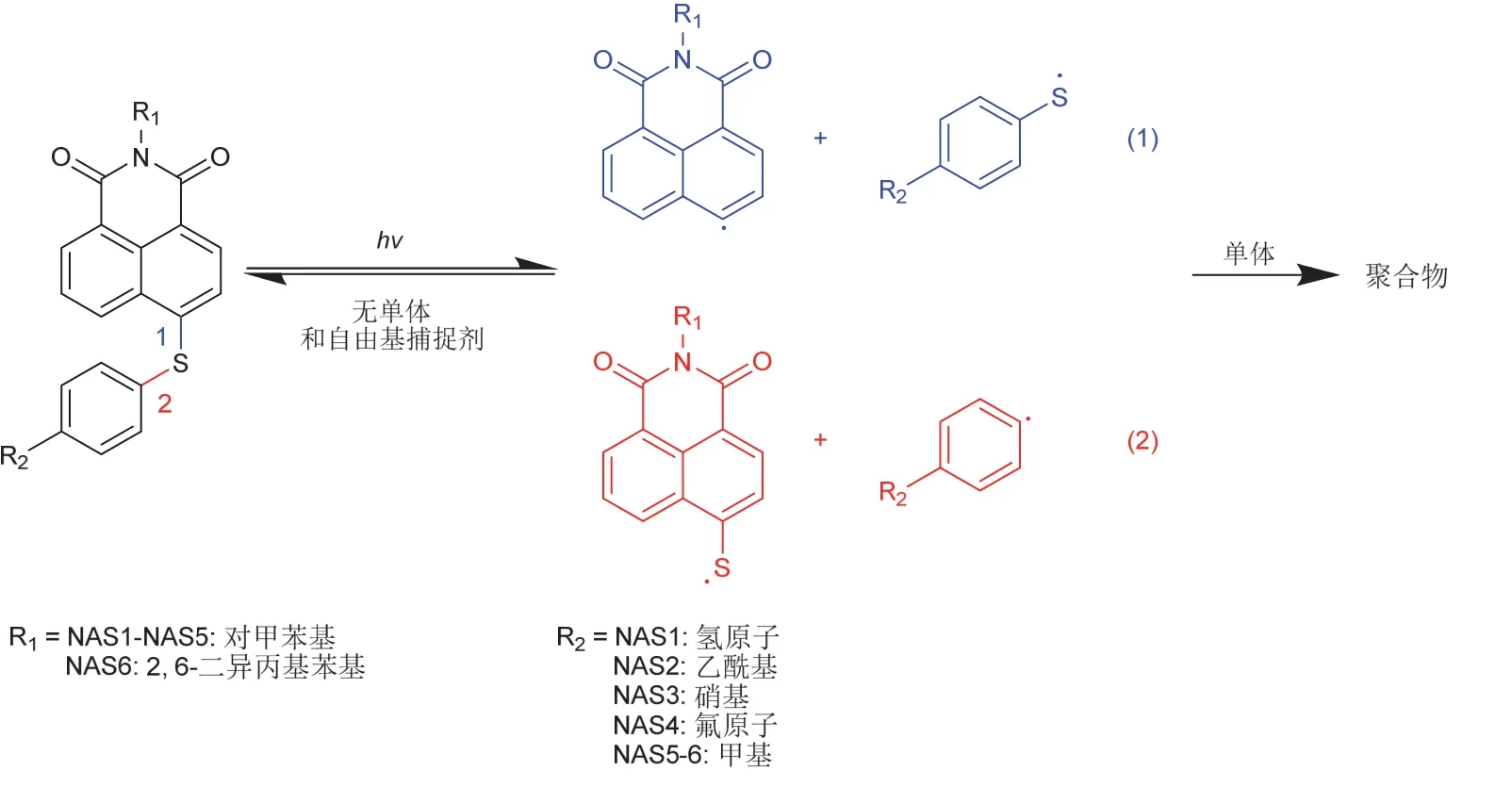
图9 推测的NASs光解机理
2.1.2 紫外光引发剂的改性
Breloy等[33]对原本用于近紫外光聚合的硫杂蒽酮进行改性,得到单胺基或二胺基取代的硫杂蒽酮衍生物,见图10中的TX1和TX2。胺基的存在使得TX1–2能以单组分的形式,在可见光下通过分子间的电子/质子转移来引发自由基聚合(图10),还能作为电子给体与(MePh)2I+作用,或电子供体与MDEA相互作用,表现出更高的引发效率。此外,这些硫杂蒽酮衍生物还可用于硫醇-烯的点击反应,是一种通用性较强的光引发剂。另外,由TX2得到的互穿聚合物网络有着支持活细胞增殖的能力,表明TX2几乎没有生物毒性,使其在食品包装和生物医药涂层中具有广阔的应用前景。
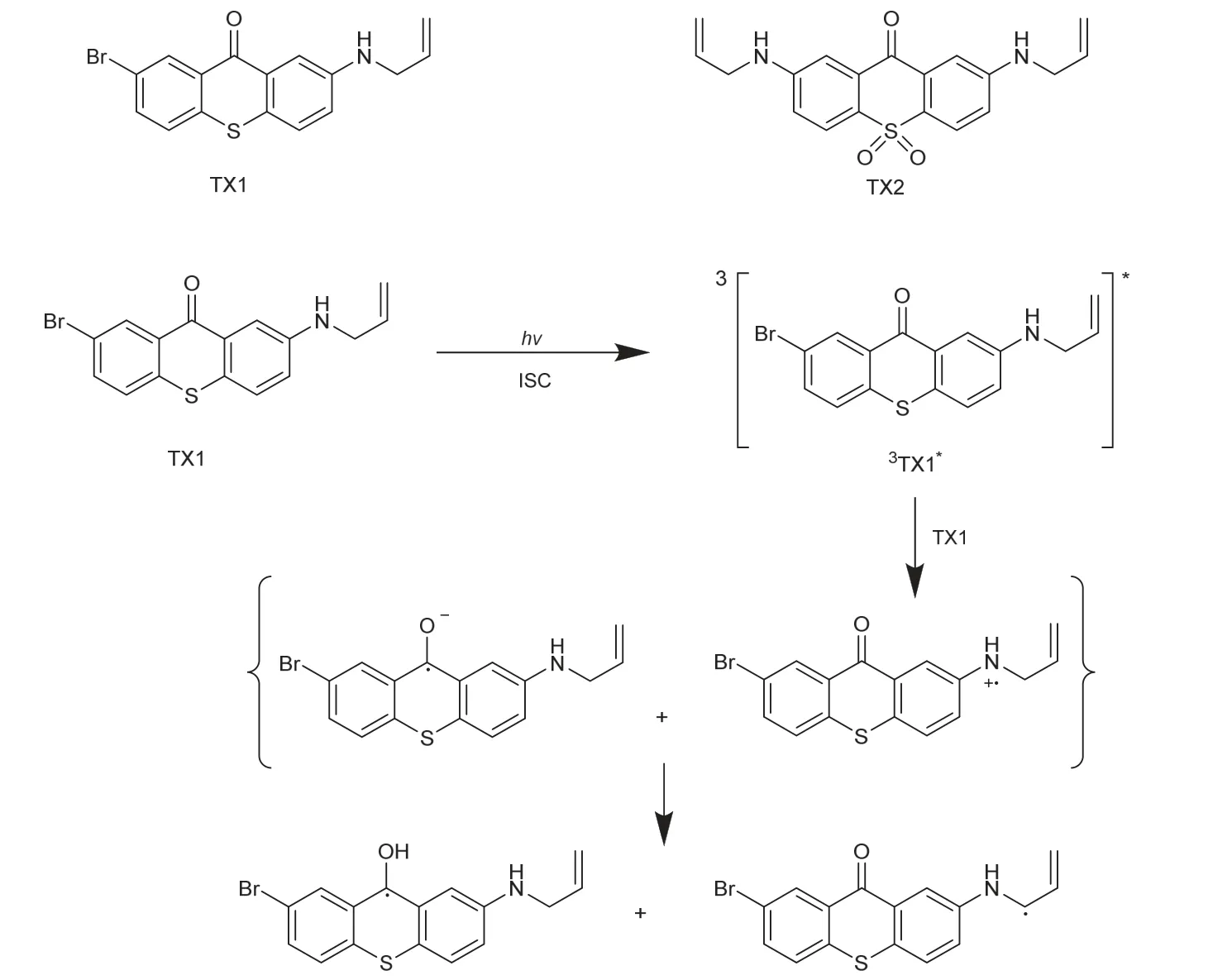
图10 硫杂蒽酮衍生物及TX1光解机理[33]
2.2 水性光引发剂
提高光引发剂水溶性最常用的方法是向原有光引发剂的结构引入季铵盐、羧酸盐等水溶性基团,这也是商业化水性光引发剂的主要设计思路。现阶段更为有趣的方法是利用超分子作用来提升光引发剂的亲水性。β-环糊精一种环状寡糖,分子外围的多个羟基使分子外表面具有亲水性,而环糊精的空腔则是疏水性的,能通过主客体相互作用,与大小适宜的脂溶性光引发剂形成超分子。Li等[34]利用这种方法制备了以1-羟基环己基苯基甲酮(HCPK)为客体分子、胺基修饰的β-环糊精衍生物CD-A为主体的水溶性超分子光引发剂HCPK-CD (图11)。由于环糊精上的胺基取代基在中性条件下占据了疏水空腔较窄侧的空间,此时HCPK进入空腔较浅。而当pH由中性变为酸性时,质子化的胺基从空腔中撤出一部分,增加了HCPK在疏水空腔所占有的体积,如图12所示。这种结构变化机制赋予了HCPK-CD随pH变化而改变的引发能力:与中性的聚合体系相比,酸性体系中,由于质子化胺基的撤出,HCPK与CD的空腔结合更为紧密,包合物的稳定性提升,使HCPK-CD在酸性条件下能够更快地引发体系聚合。此外pH还能调节聚合物表面形貌:酸性条件下超分子光引发剂的亲水性有所提升,得到的聚合物膜的孔隙率降低,膜的表面更加平整。
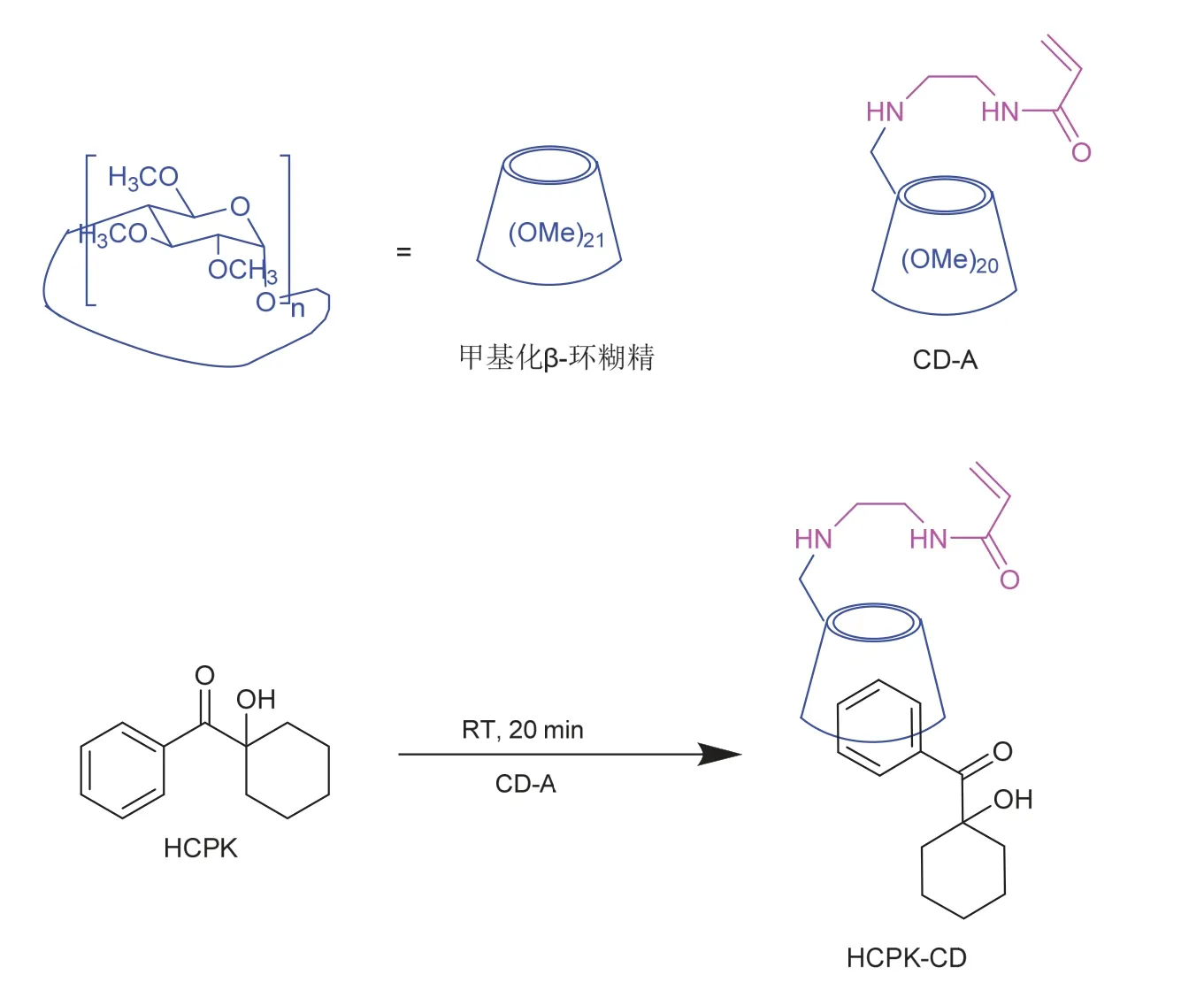
图11 HPCK-CD的制备方法[34]
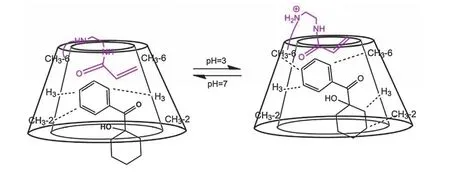
图12 HCPK-CD在不同pH下可能的结合方式[34]
2.3 大分子光引发剂
大分子光引发剂由于其分子量较大,很难从聚合物材料中迁移出来,这极大地降低了引发剂对环境的污染和人体的伤害,因此对大分子光引发剂的研发方兴未艾。制备大分子光引发剂(MPI)的方法主要有通过含羟基的小分子光引发剂与单体的共聚或缩聚反应,得到聚合物主链上有光引发剂结构的MPI;还可对已有的聚合物的侧链改性或将含有不饱和官能团的小分子光引发剂与其他单体共聚,得到侧链具有引发性能的MPI。
木质素(Lignin)是一类复杂的酚类聚合物,在植物中广泛存在,极易从自然界获得。Liu等[35]以木质素为骨架,利用其结构中的酚羟基的反应活性,在木质素骨架上接枝了提高水溶性的聚(乙二醇)(PEG)和起到光引发功能的2959结构(图13),得到一种大分子光引发剂L-PEG-2959。在紫外光的照射下,L-PEG-2959能有效引发甲基丙烯酸缩水甘油酯与明胶的光交联,形成一种有着三维网络的杂化水凝胶。由于大分子木质素结构的引入,光引发剂的迁移能力与2959相比显著下降,所得的杂化水凝胶也有较高的生物安全性。
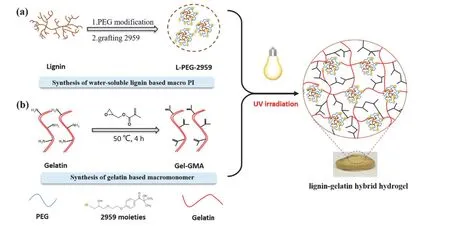
图13 木质素-明胶杂化水凝胶的制备[35]
3 结语
光聚合技术开发于上世纪70年代,针对光引发剂的研究在过去的几十年中取得了长足发展:光引发剂所匹配的光源波长迈进可见光,不再局限于紫外区域;通过引入离子盐或超分子作用,光引发剂能够在水体系保持良好的引发性能;针对小分子光引发剂易从体系中迁移的问题,大分子光引发剂应运而生。目前,该领域仍有许多难关尚待攻克:多数长波长光引发剂具有较深的颜色,阻碍了其在深层固化和无色材料中的应用,需开发无色或具有光漂白能力的长波长光引发剂;氧气对于自由基聚合有较强的抑制作用,需设计开发抗氧阻聚的光引发剂以简化生产工艺,降低生产成本;许多光引发剂的合成路线繁复,如何设计制备方法简单或能直接以天然产物为原料的光引发剂也是一个亟待解决的难题。总的来说,光引发剂的研究将不仅仅局限于实现优异的引发性能,随着研究的不断深入,开发环保、多功能的光引发剂将成为这一领域的发展方向。
