基于重启随机游走算法的生物信息学关联预测模型
2021-07-05张铭文刘志豪卢星辰
张铭文 刘志豪 卢星辰
(曲阜师范大学网络空间安全学院 山东省济宁市 273100)
1 引言
miRNA 是在真核生物中发现的一类内源性的具有调控功能的非编码 RNA, 其大小长约 20~25 个核苷酸。在医学界,研究人员主要利用生物实验的相关方法去探究 miRNA 和疾病之间的关系。但是,生物实验方法通常具有耗时长、费用高和盲目性大等缺点。因此,借助预测疾病‐miRNA 之间潜在关联的计算模型,利用各种已经验证的生物学数据,可以挖掘出二者之间关联概率得分最高的疾病‐miRNA 对,再针对性地进行生物实验,便可以有效降低实验时间和实验成本。因此,探究更为准确的疾‐miRNA 之间的关联关系,将不仅有利于人类理解疾病的发病机制,为预防和诊治疾病提供新的思路[1],而且可以反哺医学研究,为准确开展生物实验提供可靠指引。科学研究表明,功能相似的 miRNA 在调控疾病方面也比较相似,反之亦然。通过测量相似度的方法来预测 miRNA‐疾病的关联,作为较早的预测思路之一, 现如今依然有着良好的发展[2‐6]。2009年,Jiang 等人[7]通过构建疾病的表型相似网络、miRNA功能相关网络 和疾病的表型‐miRNA 网络,预测得到了疾病‐miRNA之间关联关系。该方法首次基于miRNA 的相关信息和疾病的表型信息,构建了能够预测疾病和 miRNA 之间关联关系的网络模型,为生物信息学的未来研究工作提供了新的思路。然而,该方法也存在一些不足之处,为了改进不足之处,Jiang 等人[8]在2010年再次提出了一种融合基因组数据的预测方法,利用朴素贝叶斯模型实现了多种来源数据的融合,构建了基于基因之间功能关系的信息模型,实验取得了较好的预测结果。2013年,在疾病相似性网络上,Chen 等人[9]应用随机游走算法实现了 miRNA‐疾病关联预测,此后,Liu 等人[10]融合疾病的语义相似信息、miRNA 的功能相似信息和 miRNA 与疾病之间已知的关联信息,构 建了多源异构网络,继而利用随机游走算法预测疾病‐miRNA 之间的关联关系。实验表明,该方法可以预测未知miRNA 关联信息的新疾病。值得介绍的是,多 源异构网络的应用对后续 miRNA‐疾病关联的预测研究具有巨大的借鉴价值。2018年,Chen 等人[5]提出了一种基于归纳矩阵填充的算法来预测 miRNA‐疾病之间的关联。首先利用 miRNA的功能相似性、疾病语义相似性和高斯相似 性来计算 miRNA 和疾病各自的集成相似性,然后采用归纳矩阵填充的方法从中归纳出miRNA 和疾病的有效特征信息,并用来填充 miRNA‐疾病关联矩阵中的缺失信息。2019年,Chen 等人[4]提出一种基于二分异构网络联合近邻的 miRNA‐疾病关联预测算法,根据网络上简单路径的数量计算出来 miRNA 和疾病各个结点的初始关联分数,而疾病和miRNA 各自的次级空间关联得分则由其自身的相似性 网络和初始关联得分计算得出,最后将二者的次级空间得分加以整合得到了最终的 miRNA‐疾病关联预测得分。2020年,Wang 等人[6]提出了一种基于数据融合的miRNA‐疾病关联预测算法,通过将miRNA功能相似性网络、基因功能相似性网络和疾病语义相似性网络融合在一起来对 miRNA 和疾病之间的关联关系进行预测,弥补了以往因为使用蛋白质之间相互作用网络而造成假阳性结果的不足。但以往的研究都忽略了数据自身的拓扑结构,因此本文采用重启随机游走的算法充分挖掘了数据的拓扑网络相似性,使得预测模型的准确性得到进一步的提高。RWRCMF 模型流程图如图1所示。
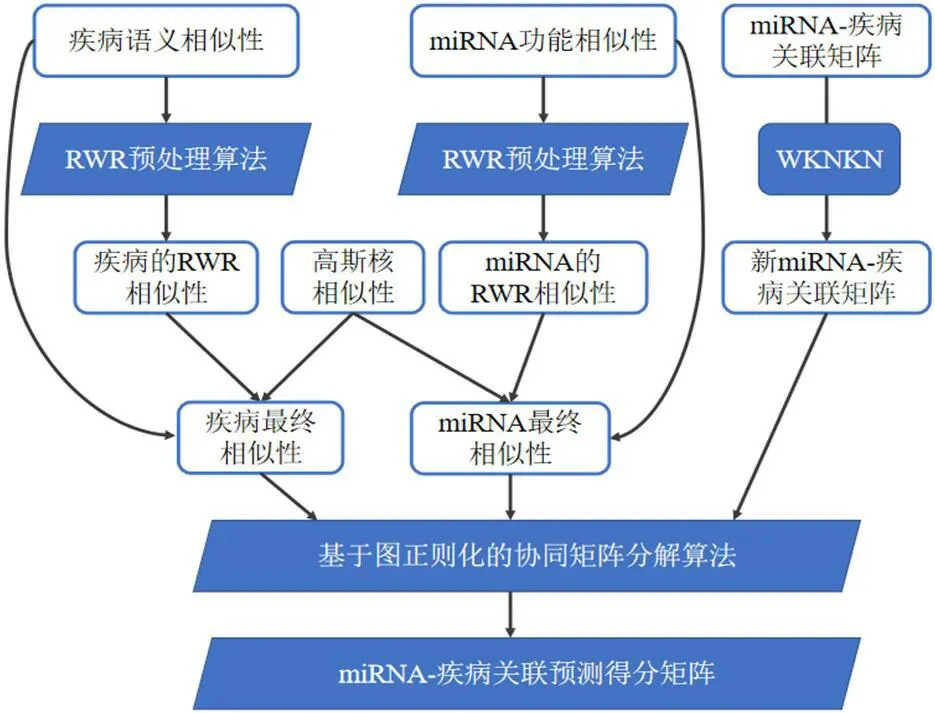
图1:RWRCMF 模型流程图
2 数据获取及预处理
2.1 已知的miRNA-疾病关联
现有的HMDDv2.0[11]数据库中包含了383 个疾病和495 个miRNA 之间5430 已被验证的关联信息。本文利用5430 个已知关联构建出一个邻接矩阵用来存储这些数据,其中n 表示miRNA 的个数,m 表示疾病的个数。经实验验证后的miRNA mi如果和疾病dj有关联,则矩阵元素Yij为1,否则为0。
2.2 miRNA的功能相似性
研究发现,那些功能相似性的miRNA 往往更可能与相似的疾病有关联,反之亦然[12‐13]。借助Wang 等人[14]的研究,本文得到了miRNA 之间的功能相似性分数,并以此为据构造出了miRNA的功能相似性矩阵矩阵中的各个元素值代表了miRNA mi和mj二者的功能相似性得分。
2.3 疾病语义相似性
本文根据Wang 等人[14]提出的层级有向无环图(DAG)计算得出了疾病的语义相似性。疾病之间的DAG 图从MeSH 数据库中下载。DAGd=(d, Td, Ed)表示疾病d 的DAG 图,其中Td表示疾病的集合,Ed表示DAG 图中互相连接关系的结合。由此,本文将根据DAG 图按照以下公式计算疾病D 的语义值:

其中,DD(d)表示疾病d’对疾病d 的语义贡献值,如公式(2)所示,表示语义贡献因子,由Wang 等人[14]文献可知=0.5。

因此,根据两个疾病在DAG 图上重复越多,则其彼此之间相似性更大的理论假设,本文由公式(3)计算得到疾病di和dj之间的语义相似性,并构建出了疾病的语义相似性矩阵其中矩阵的每个元素SD(di, dj)表示为疾病di和dj之间的语义相似性得分。
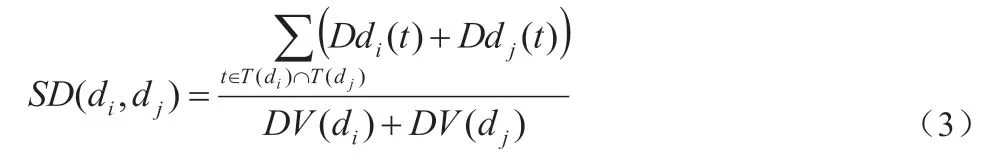
2.4 miRNA和疾病的拓扑相似性
为了提高预测结果的准确性,本文借助公式(4)和(5)在相似网络上运用重启随机游走算法(RWR)得到了miRNA 和疾病各自的拓扑相似性。
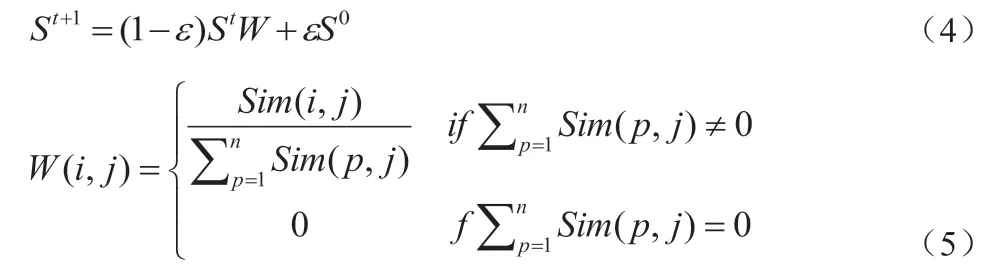
在经过数次迭代后,我们得到了一个稳态矩阵S∞,当Sim 的值为矩阵SM 时,所得的S∞=SRM,表示miRNA 的拓扑网络相似性矩阵;当Sim 的值为矩阵SD 时,所得的S∞=SRD,表示疾病的拓扑网络相似性矩阵。
2.5 高斯核相互作用谱相似性
以往许多对miRNA‐疾病之间关联预测的模型都运用了高斯核相互作用谱相似性来度量疾病和miRNA 各自的相似性,并达到了较好的预测结果,本文用IP(di)和IP(mj)分别表示疾病di和miRNA mj的相互作用谱,并根据公式(6)计算疾病di和dj之间的高斯核相互作用谱相似性。
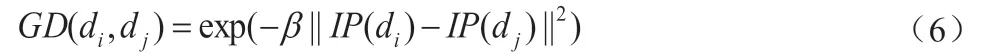
类似的,本文借助公式(7)计算miRNA mi和mj的高斯核相互作用谱相似性。
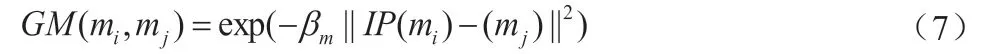
其中的核带宽βm、βd由如下公式定义,和为原始带宽。
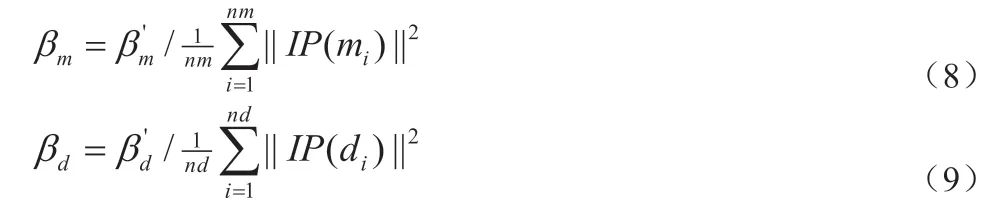
因此,疾病和miRNA 的高斯核相互作用谱相似性分别用矩阵GD、GM 来表示。
2.6 此疾病语义相似性
根据上述各种方法完成了miRNA 和疾病相似性度量,并将这些相似性进行整合得到了最终的miRNA 相似性和疾病相似性,其中miRNA 的功能相似性矩阵SMM 由公式(10)计算得来,疾病的语义相似性矩阵SDD 由公式(11)计算得来,元素SMM(mi, mj)表示miRNA mi和mj的相似性,元素SDD(di, dj)表示疾病di和dj的相似性。

2.7 加权K近邻(WKNKN)预处理
将n 个miRNA 和m 个疾病分别用集合M={m1,m2,…mn}和D={d1,d2,…dn}来表示。已知邻接矩阵表示现有数据库中的miRNA 和人类之间的疾病关联,但矩阵Y 中的元素过于稀疏,因此利用加权K 近邻(WKNKN)算法对矩阵Y 进行预处理,使得矩阵中的0 元素会被一个0 到1 之间的约数所替换。WKNKN 的具体步骤如下:
首先,根据miRNA mq与其他K 个最近邻miRNA 的相似性和所对应的K 相互作用关系,得到下列相互作用谱:

公式中,m1到mk是将与mq相似性进行降序排列之后所得的miRNA。表示权重参数,且mi和mq之间相似的分越高,权重参数值越大。是衰减控制项,是用来归一化的参数。
同理可得,疾病dp和其K 个近邻疾病的相似性以及对应的K相互作用谱,得到了如下疾病作用谱。

二公式中dl到dk是由各自与dp的相似性按照降序排列之后的疾病,表示权重参数,且dj和dp之间相似性得分越高则权重就越大。类似的,是归一化参数。
最后,将关联矩阵Y 中的0 元素用Ym和Yd的平均值替换下来,并按照公式(12)对原始矩阵Y 进行更新。

3 改进协同矩阵分解
3.1 标准协同矩阵分解
此Shen 等人[15]将协同矩阵分解模型用在了miRNA‐疾病关联预测问题上,并达到了预期效果。CMF 目标函数如下:
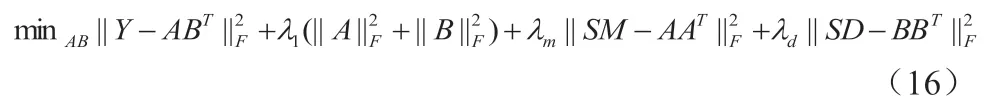
3.2 基于图拉普拉斯正则化的协同矩阵分解
处为了降低数据的过拟合问题,提升模型在预测miRNA‐疾病关联过程中的准确性,本文在原有矩阵分解的基础上引入图拉普拉斯正则化,并构造出了如下目标函数:
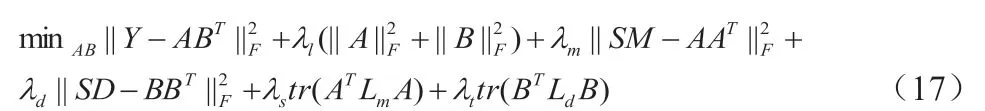
3.2.1 初始化A、B
在miRNA‐疾病关联矩阵Y 进行预处理的过程中,本文采用奇异值分解(SVD)的方式初始化矩阵A、B,过程如下:

3.2.2 优化过程
本文采用最小二乘法对矩阵A、B 进行迭代求解。首先,将目标函数用L 来表示,然后令求得更新之后的迭代规则,此时A、B 将按如下公式迭代更新直到收敛。

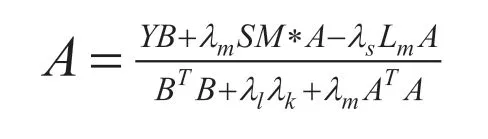
4 实验结果分析
4.1 性能评估
为了进一步对重启随机游走算法改进后的协同矩阵矩阵分解预测模型(RWRCMF)进行预测性能的评估,本文将RWRCMF 与三个主流模型ICFMDA[16]、SACMDA[17]、IMCMDA[18]进行对比,模型的评估标准使用机器学习方法中的五折交叉验证和留一交叉验证。
降根据留一交叉验证的结果,本文绘制了对应的接收器操作特性(ROC)曲线,因此可以通过计算得出ROC曲线下的面积(AUC值)来评估预测模型的性能。AUC 值的大小介于0 到1 之间,并且模型的预测性能和AUC 值的高低成正比。由图2 可知,RWRCMF、IMCMDA、ICFMDA 和SACMDA 所对应的AUC 值分别为:0.9506、0.8384、0.9072、和.8777,显然RWRCMF 预测模型在上述方法中获得了最佳的预测表现。
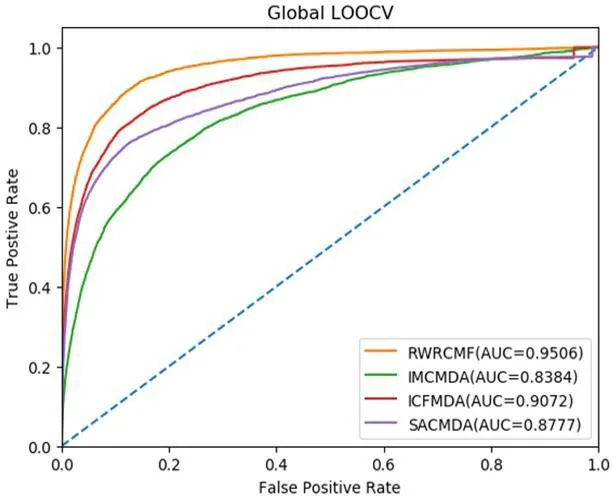
图2:RWRCMF、IMDMDA、ICFMDA 和SACMDA 的留一交叉验证ROC曲线以及对应的AUC 值
在对模型的五折交叉验证过程中,首先将已知的miRNA‐疾病关联样本任意分成五部分,然后选择其中的四部分作为模型在学习过程中的训练样本,余下的作为评价模型的测试样本。类似于留一交叉验证,所有未知关联的miRNA‐疾病关联样本来作为候选样本。同时因为考虑到样本在划分过程中因为随机性而导致的偏差,本文进行了100 次的划分,也就是执行了100 次的五折交叉验证,最终得到了各个模型的ROC 曲线和对应的AUC 值。图3 结果表明RWRCMF 模型的预测效果最好,其AUC 值为0.9468,而IMCMDA、ICFMDA 和SACMDA 的AUC 值分别为0.8330、0.9046和0.8773。
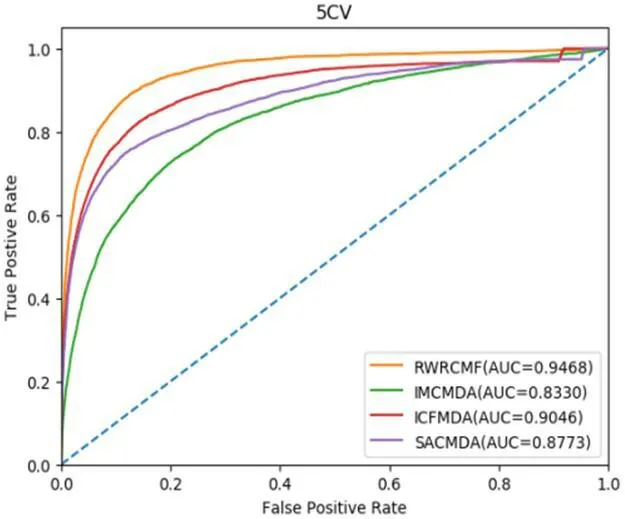
图3:RWRCMF、IMDMDA、ICFMDA 和SACMDA 的五折交叉验证ROC曲线以及对应的AUC 值
5 总结
越来越多的研究发现miRNA 的异常表达影响着人类复杂疾病的产生和发展,传统实验的方法探究miRNA 和疾病之间的关联有着耗时长、成本高的缺点,因此开发更高效的计算模型用于miRNA‐疾病的潜在关联识别显得尤为重要。在本文中,我们提出了一个基于重启随机游走算法改进后的协同矩阵分解预测模型(RWRCMF)用于识别miRNA‐疾病之间的新关联,通过重启随机游走算法处理疾病和miRNA 的相似性数据,并整合高斯核谱相似性来作为最终的数据集。借助协同矩阵分解中的目标函数获得了miRNA‐疾病的关联得分矩阵,并在实验评估环节采用留一交叉验证和五折交叉验证横向对比了RWRCMF 与以往模型的预测准确性,证实了RWRCMF 在miRNA‐疾病关联预测方面有着较为出色的表现。
