有机发光材料中间体5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬的合成工艺
2021-06-27刘安昌李惠竹何良莉
刘安昌,李 琪,邓 三,李惠竹,何良莉
武汉工程大学化工与制药学院,湖北 武汉430074
化学名称5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬[1-3],英文名:5,10-Dihydro-5-phenylbenz[b]indolo[2,3-d][1]benzazepine,分子量358.43,CAS号1799295-84-3,结构式如下:
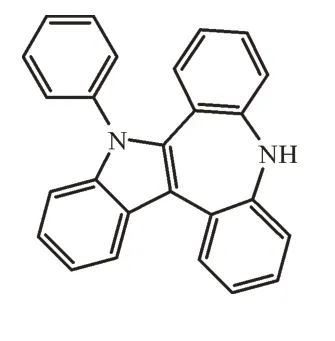
5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬是有机发光材料的重要中间体,目前国内报道很少,其合成方法是由中间体5,11-二氢-10H-二苯并[d,f]氮杂䓬和N,N-二苯基肼在乙酸条件下环化而成[4-6],其合成路线如图1所示。
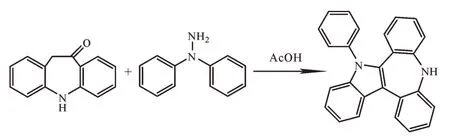
图1 5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬的合成路线Fig.1 Synthetic routeof 5,10-dihydro5-phenyl benzo[b]indoline[2,3-d][1]benzoazapine
其中,1,1-二苯基肼的合成是在二苯胺在亚硝酸钠的作用下,生成二苯基亚硝胺,后经二氧化硫脲等还原制得[7-9]。其合成路线如图2所示。
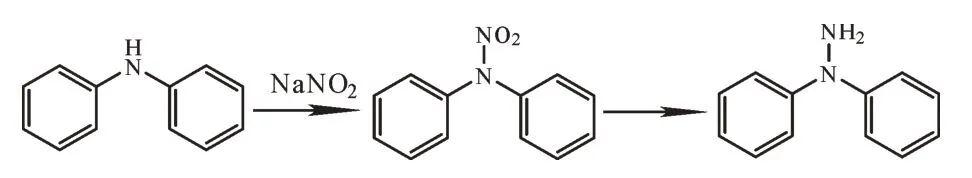
图2 1,1-二苯基肼的合成路线Fig.2 Synthesis route of 1,1-diphenylhydrazine
5,11-二氢-10H-二苯并[d,f]氮杂䓬的合成路线主要有两条。路线一[10-12]:以5-H-二苯并[b,f]氮杂䓬为起始原料,经酰胺化,mCPBA氧化得到1-(1Ah-二苯并[b,f]环氧乙烯[2,3-d]氮杂䓬-6(10Bh)-基)乙酮,然后在碘化锂的作用下,环氧基开环,乙酰基去保护得到目的产物,该路线原料5,11-二氢-10H-二苯并[d,f]氮杂䓬不易得,难以实现工业化。合成路线一如图3所示。
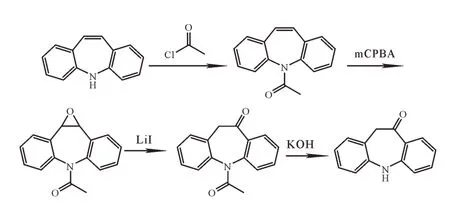
图3 5,11-二氢-10H-二苯并[d,f]氮杂䓬合成路线一Fig.3 Synthetic route Iof 5,11-dihydro-10H-dibenzo[d,f]azapine
合成路线二[13-15]:以苯胺,2-溴苯乙酸为原料,在铜粉得催化下得到2-(2-(苯基氨基)苯基)乙酸;然后与乙醇酯化,生成2-(2-(苯基氨基)苯基)乙酸乙酯;后与对甲基苯磺酰氯反应保护氨基生成2-[2-(N-苯基-N-对甲苯磺酰胺基)苯基]乙酸乙酯;其后在三氯化铝作用下分子内环化生成5,11-二氢-10H-二苯并[d,f]氮杂䓬。该合成路线较短,原料也易得,适合工业化生产。合成路线二如图4所示。

图4 5,11-二氢-10H-二苯并[d,f]氮杂䓬合成路线二Fig.4 Synthetic route IIof 5,11-dihydro-10H-dibenzo[d,f]azapine
1 试验部分
1.1 仪器和试剂
RY-1熔点仪,ZF-20D暗箱式紫外分析仪,Mercury300核磁共振仪(溶剂为CDCl3,TMS为内标),温度计未校正,N,N-二苯基肼为实验室自制,其余所用试剂和溶剂均为试剂级。
1.2 1,1-二苯基肼的合成
在装有搅拌棒滴液漏斗得500 mL的四口烧瓶中加入33.8 g(0.2 mol)二苯胺,30 g(0.5 mol)冰醋酸和200 mL甲醇,搅拌溶解后,冷却至15℃,滴加27.6 g(0.4 mol)亚硝酸钠的100 mL水溶液,于15~20℃反应8 h后,薄层色谱分析(thin-layer chromatography,TLC)检测反应完全,加入50 g(1.25 mol)氢氧化钠的200 mL水溶液,43.2 g(0.4 mol)二氧化硫脲加入反应瓶中,于38~45℃继续搅拌3 h,反应结束后蒸掉甲醇,用甲苯萃取,稀盐酸洗涤,然后水洗,减压浓缩,得到粗品用乙醇重结晶29.7 g,收率81%。
1.3 2-[2-(苯基氨基)苯基]乙酸的合成
将9.3 g(0.1 mol)苯胺,10.8 g(0.05 mol)2-溴苯乙酸,17.3 g(0.125 mol)碳酸钾,2 mL吡啶和0.4 g铜粉倒入250 mL三口瓶中,在110~120℃温度下,搅拌10 h。反应完毕,冷却,加入150 mL水,过滤除去铜粉,然后用30 mL正己烷萃取,得到的水相用盐酸酸化至pH 1~2,有大量的固体析出。过滤,水洗干燥后得9.6 g粗品,收率85%。文献[5]收率83%。
1.4 2-[2-(苯基氨基)苯基]乙酸乙酯的合成
在250 mL的反应瓶中加入45.4 g(0.2 mol)的2-[2-(苯基氨基)苯基]乙酸,无水乙醇150 mL,浓硫酸4 mL,搅拌加热至回流反应10 h,TLC跟踪监测。反应完成后,将反应物冷却蒸掉乙醇,残余物加入150 mL水稀释,用质量分数30%碳酸钠水溶液中和,后用100 mL乙酸乙酯萃取3次,合并有机相,浓缩得45.9 g淡黄色油状,收率90%。文献[5]收率82%。
1.5 2-[2-(N-苯基-N-对甲苯磺酰胺基)苯基]乙酸乙酯的合成
在装有搅拌器,回流冷凝管的500 mL四口反应瓶中,加入25.5 g(0.1 mol)2-(2-(苯基氨基)苯基)乙酸乙酯,23.7 g(0.125 mol)对甲苯磺酰氯,9.8 g(0.125 mol)吡啶和250 mL氯仿,反应混合物在室温下搅拌2 h,后加热回流反应3 h,反应完成后冷却至室温,加入100 mL水,分出水层,用50 mL氯仿萃取3次,合并有机相,有机相分别用质量分数5%的盐酸洗涤,饱和碳酸氢钠溶液和水洗涤,浓缩,粗品用丙酮重结晶,得35.2 g白色固体,收率85%。文献[5]收率84%,熔点152~154℃。
1.6 5,11-二氢-10H-二苯并[d,f]氮杂䓬的合成
在一个500 mL的四口反应瓶中,加入26.7 g(0.2 mol)无水AlCl3,150 mL二氯甲烷搅拌溶解,滴加40.9 g(0.1 mol)2-[2-(N-苯基-N-对甲苯磺酰胺基)苯基]乙酸乙酯溶于50 mL二氯甲烷的溶液,滴加时间约30 min,反应回流6 h后冷却,将反应液倒入200 mL,质量分数5%的稀盐酸中,用150 mL二氯甲烷萃取3次后,分别用质量分数10%碳酸钠水溶液和水洗涤,浓缩,粗品用苯重结晶16.7 g,收率80%。熔点:142~145℃。
2 结果与讨论
在氮气保护下,将20.9 g(0.1 mol)5,11-二氢-10H-二苯并[d,f]氮杂䓬,20.2 g(0.11 mol)1,1-二苯基肼,100 mL乙酸加入到250 mL的反应瓶中,于120℃搅拌反应12 h,反应完成后,将反应物倒入200 mL水中,用二氯甲烷萃取,有机层用无水硫酸镁干燥过滤,去除溶剂后,产品重结晶得27.2 g的收率74%。较文献[1]收率73%略高。熔点为:145~146℃,并通过1H-NMR(CDCl3)表征:δ6.65~6.80(m,2H),6.90~6.94(m,2H),7.10(t,1H),7.20~7.24(m,7H),7.32~7.37(m,3H),7.61(b,1H),7.85(d,1H),8.01(d,1H)与5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬相符,证明已成功合成了5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬。
3 结 论
以苯胺、2-溴苯乙酸为原料,合成2-(2-(苯基氨基)苯基)乙酸;然后在硫酸的催化下与乙醇酯化,生成2-(2-(苯基氨基)苯基)乙酸乙酯;后与对甲基苯磺酰氯反应保护氨基生成2-(2-(N-苯基-N-对甲苯磺酰胺基)苯基)乙酸乙酯;其后在三氯化铝作用下发生分子内环化生成5,11-二氢-10H-二苯并[d,f]氮杂䓬;最后在乙酸中与N,N-二苯基肼反应生成目标产物5,10-二氢-5-苯基苯并[b]吲哚并[2,3-d][1]苯并氮杂䓬。该合成工艺简单,原料易得,适合工业化生产。
