超高效液相色谱-高分辨质谱定量检测蜂毒衍生品中蜂毒肽的含量
2021-06-21蔡冬梅张玉豪孙菲菲周金慧杨宇晖杨术鹏
蔡冬梅 傅 怡 张玉豪 孙菲菲 周金慧 杨宇晖 杨术鹏, 李 熠
(1.中国农业科学院蜜蜂研究所,农业农村部蜂产品质量安全控制重点实验室,北京100093;2.安徽农业大学动物科技学院,合肥230036;3.中国农业科学院农产品加工研究所,北京100193)
蜂毒是由蜜蜂工蜂毒腺和副腺分泌的毒液,它是蜜蜂蜇刺敌害时释放的一种生物武器。蜂毒中含有丰富的酶、多肽、生物胺、激素、有机酸、氨基酸等物质,其中蜂毒肽(蜂毒溶血肽)是蜂毒中主要成分,占蜂毒干重的50%以上[1]。蜂毒肽是由24个氨基酸组成的小肽类物质,其氨基酸序列为GIGAVLKVLTTGLPALI SWIKRKRQQ,其中6个亲水性氨基酸残基在C端,20个疏水性氨基酸残基在N端,呈α-螺旋构象[2]。蜂毒肽具有广泛且强烈的生物学活性,如抗菌、抗炎、镇痛、免疫调节、抗病毒和抗肿瘤等功效[3~6]。目前,我国和全球多个国家均已批准蜂毒肽作为上市药物,用于治疗风湿性疾病和周围神经炎。近年来研究发现,蜂毒肽在皮肤保养和皮肤病防治方面疗效确切,它能刺激肌肤胶原蛋白产生,抚平细纹,恢复弹性,从而使得蜂毒肽成为护肤品行业的新宠[7]。目前,国内外已经有众多企业开展了蜂毒肽在护肤品中应用探索,并在市场上推出了各式各样的护肤产品,如蜂毒保湿水、蜂毒护肤霜、蜂毒面膜等。蜂毒产量非常稀少,从而导致蜂毒肽原料价格非常昂贵。护肤品中添加蜂毒肽会显著增加生产成本,其价格显著高于普通护肤品。当前,我国并未颁布有关护肤品等产品中蜂毒肽的含量测定标准,不同企业的生产方式和质控标准并不统一,从而导致市场上蜂毒肽类护肤品的质量参差不齐,甚至出现了假冒伪劣的现象。因此,对蜂毒肽的准确定量分析方法的开发已经成为养蜂业和护肤行业的共识。但由于蜂毒肽众多衍生产品中蜂毒肽含量很低,仅处于痕量水平,而其基质又十分复杂,亟需开发一个可精确定量蜂毒肽含量的方法。
有关蜂毒肽的检测方法主要是免疫分析和仪器分析。抗原抗体特异性识别是免疫分析方法的基础,KING等[8]通过免疫小鼠,获得蜂毒肽的多克隆抗体和单克隆抗体。但是作者并未基于获得抗体开展定量分析方法开发。尽管免疫分析方法具有简单、方便等优点,但是由于抗体存在制备周期长、定量方法结果准确度和稳定性差等弊端,限制了其在蜂毒肽定量中的应用。基于色谱的仪器分析在蜂毒肽定量分析方面有广泛报道,也是当前蜂毒肽定量的主要检测方法。早期报道的仪器分析方法主要基于高效液相色谱结合紫外检测器(HPLC-UV),但是这类方法存在定量限高、准确度和精密度有限以及抗干扰能力差等弊端[9]。随着液相色谱串联质谱(LC-MS)技术的成熟与普及,LC-MS已经成为近10年来蜂毒肽的主要检测方法。液相串联三重四极杆质谱(LC-MS/MS)、液相串联离子阱质谱(LC-IT)以及液相色谱串联高分辨质谱(LC-HRMS)也相继应用到各种基质中蜂毒肽的定量[10~14]。由于蜂毒肽的化学结构中含有24个氨基酸,采用LCMS检测时,其离子化效率有限,从而导致建立方法的灵敏度依然有限[12~13]。此外,已报道的LCMS方法主要采用外标法进行定量分析,难以适用于众多的蜂毒衍生品中蜂毒肽的准确定量[10~11,13]。近年来,随着蛋白质组学和肽段合成技术的快速发展,基于LC-MS的靶向蛋白质定量技术取得了巨大突破。与直接检测小肽相比,基于蛋白酶水解策略结合同位素标记特征性肽段的定量技术在方法的灵敏度、准确度、稳定性和方法实用性等方面具有显著优势。本研究拟采用该策略并结合UHPLCHRMS开展蜂毒衍生品中蜂毒肽的定量分析方法开发和有效性评价,为保护蜂毒肽消费者的权利提供方法支持,同时为后续蜂毒肽行业标准的制定提供方法参考。
一、材料与方法
(一)仪器与试剂
1.仪器与设备。超高效液相色谱串联四极杆/高分辨静电轨道阱质谱(UHPLC Q Executive Plus,美国Thermo Fisher Scientific公司);台式低温离心机(Microfuge 22R Centrifuge,美国Beckman Coulter公司);全波长酶标仪(Multiskan GO,美国Thermo Fisher Scientific公司);电子分析天平(PL203,德国Mettlertoledo公司);蒸发浓缩仪(Speed-Vacsvstem,RVC2-18, 德国MarinChrist公司);超纯水机(Milli-Q,美国Millipore公司);超低温冰箱(MDF-U3286S,日本SANYO公司)。
2.试剂与标准品。碳酸氢铵(NH4HCO3)购买于Solarbio公司;磷酸缓冲盐溶液(phosphate buffer saline,PBS)、 二硫苏糖醇(DL-dithiothreitol,DTT)、 碘乙酰胺(iodoacetamide,IAA) 购于Sigma-Aldrich公司; 三氟乙酸(trifluoroacetic acid,TFA)购于J.T.Baker公司;质谱级胰蛋白酶(trypsin) 购 自Promega(Madison,WI,USA) 公 司 ,甲酸(formic acid,FA)、 甲醇(methanol)、 乙腈(acetonitrile,ACN)购买于Fisher公司;C18除盐洗脱柱购于3M公司(Minnesota Mining and Manufacturing)。
特征肽段VLTTGLPALISWIK;稳定同位素标记的内标(SIL-IS)肽段VLTTGLPALISWIK*(K*表示赖氨酸中的碳置换为13C,氮置换为15N)由南京源肽生物科技有限公司合成,纯度≥98%,储存在-20℃备用。
(二)样本前处理方法
1.溶液配制。40 mM NH4HCO3溶液:称取0.316 g NH4HCO3,用超纯水定容100 mL,4℃冰箱保存待用。100 mM DTT:称取DTT 308.5 mg,40 mMNH4HCO3溶液定容到20 mL,每管l mL分装,-20℃冰箱保存待用。100 mM IAA溶液:称取IAA 0.39 g,用40 mM的NH4HCO3溶液定容至20 mL,-20℃冰箱保存待用。活化液:500μL ACN、3μL TFA,超纯水定容至1 mL,4℃保存。平衡液:1μL TFA,超纯水定容至1 mL,存于4℃。洗脱液:800μL ACN、1μL TFA,超纯水定容至1 mL,4℃保存。
2.样本前处理方法。
(1)方法A:未酶切前处理方法。准确称取蜂毒衍生品样品1 g至10 mL离心管中,加入10 mL甲醇,涡旋溶解后,35℃氮吹至近干。加入1 mL去离子水涡旋复溶,于14 000 rpm 4℃条件下离心12 min。收集上清液于新的2 mL离心管中。向每个样品中加入200 ng/mL IS肽段进行质谱分析。(2)方法B:酶切前处理方法。准确称取蜂毒衍生品样品1 g至10 mL离心管中,加入10 mL甲醇,涡旋溶解后,35℃氮吹至近干。加入1 mL去离子水涡旋复溶,于14 000 rpm4℃条件下离心12 min。收集上清液于新的2 mL离心管中。移取蛋白质溶液200μL与800μL 40 mM NH4HCO3混合。向上述混合溶液中加入100μL 30 mMDTT溶液,在室温下反应60 min,然后再加入500μL 100 mMIAA溶液于室温下暗反应60 min。样品中加入30μL胰蛋白酶溶液,于37℃过夜酶切12 h。酶切反应完成后,加入1μL甲酸使胰蛋白酶失活,终止酶切反应,向每个样品中加入200 ng/mL IS肽段。混合均匀后使用C18柱除盐,将除盐后的肽段干燥,用100μL初始流动相复溶进入液相色谱串联质谱检测。
(三)仪器方法
1.色谱条件。色谱柱为Thermo Hypersil GOLDTMC18(2.1 mm×100 mm,1.9μm); 流动相组成:A为0.1%甲酸水溶液,B为0.1%甲酸乙腈;梯度洗脱条件为:0~1.0 min,5%流动相B;1.0~2.0 min,5%~15%流动相B;2.0~12.0 min,15%~40%流动相B;12.0~14.0 min,40%~95%流动相B;14.0~16.5 min,95%流动相B;16.5~17.5 min,95%~5%流动相B;17.5~20.0 min,5%流动相B。流速为0.30 mL/min;进样量为5.0μL;柱温为40℃。
2.质谱条件。离子源参数:鞘气的流速为38 arb;辅助气的流速为15 arb;挡锥气的流速为0 arb;电喷雾电压3.2 kV;离子导管的温度275℃;S-lens RF level设为60;离子源温度380℃;采集的模式为正离子模式下的Full MS-ddMS2;其中Full MS的具体参数设置为:分辨率为70 000;AGC Target为3e6;最大离子注入时间为200 ms;质量扫描范围为300~1 200 Da;数据类型为棒状图;dd-MS2的具体参数设置为:分辨率为17 500;AGC Target为1.6e5;最大离子注入时间为120 ms;Loop count为2;隔离窗口为2.0 Da;NCE为35;数据类型为棒状图;dd settings中,最小AGC为8.0e3;顶点触发为2~6 s;同位素排除为on;动态排除为8.0 s。
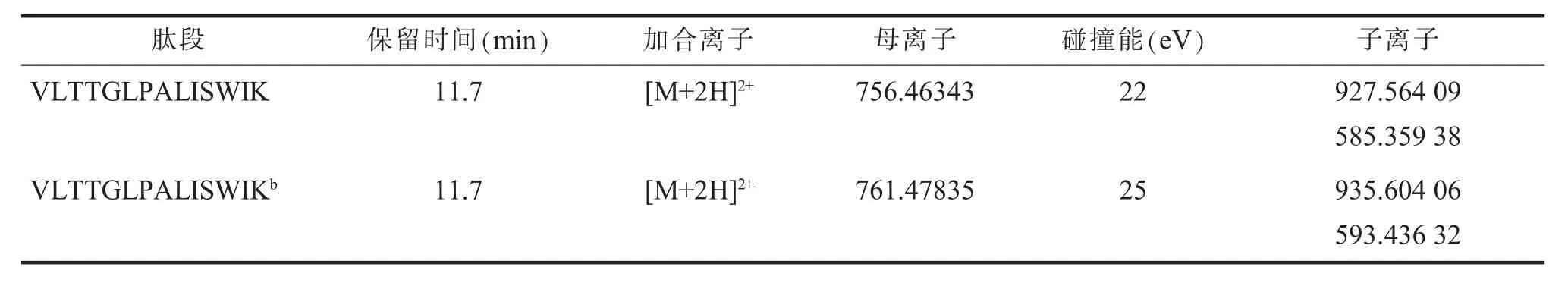
表1 蜂毒肽特征肽段及同位素内标肽段的质谱分析参数
(四)方法学验证
1.特异性。在蜂毒肽样品中和空白样品中提取筛选好的蜂毒肽特征肽段,观察目标肽段的色谱峰的信噪比,明确空白样本中是否存在干扰峰。
2.线性范围。采用内标法定量,使用初始流动相分别配制特征肽段的同位素内标标准曲线,设置特征肽段浓度点分别为3、6、15、50、150、500 ng/mL,其中每个浓度点的标准溶液中都含50 ng/mL特征肽段对应的同位素内标肽段标品。准备好样本由UHPLC-HRMS进行分析。
3.检测限、定量限。不含蜂毒肽的空白样品按照前处理方法操作后分析,将目标色谱峰的信噪比≥3时的浓度,设定为检测限(LOD);同时将信噪比≥10时的浓度设定为定量限(LOQ)。
4.准确度和精密度。本试验选择特征肽段低(LOQ)、中(2LOQ)、高(10LOQ)3个浓度水平进行回收率和精密度试验。按前处理方法操作后分析,每个浓度重复6份,连续做3 d,计算日内精密度及日间精密度,得到的结果作为整个试验方法的准确度和精密度。
二、结果与分析
(一)蜂毒肽特征肽段的筛选与确立特异性肽段的选择是液相色谱串联质谱定量靶向蛋白质方法开发的关键和难点,直接决定了后续建立方法的特异性、灵敏度和稳定性。选择特征肽段时应遵循的基本原则是,肽段中氨基酸数目以8~20为宜,不应过短或者过长;且为质谱和液相友好型;选择的肽段应具备特异性、稳定性和一定离子丰度的专属性肽段,需通过蛋白质数据库的比对;肽段中应当避免含有易于修饰的氨基酸,如天冬酰胺(N)、 谷氨酸(E)、 天冬氨酸(D)、 丝氨酸(S)、苏氨酸(T)、半胱氨酸(C)等;肽段中应避免含有胰蛋白酶漏切的位点(K或R)。通过与NCBI及UniProt数据库比对,蜂毒肽水解之后的肽段VLTTGLPALISWIK具有唯一性,且在液相色谱串联质谱上具有较好的相应值。蜂毒肽水解之后另外一条肽段GIGAVLK在搜库匹配时,不具备唯一性,且在质谱中响应较低。因此,本研究将VLTTGLPALISWIK作为蜂毒肽的特征肽段开展液相色谱串联质谱的定量分析方法开发。
(二)检测方法的灵敏度、线性范围、准确度与精密度
1.选择性。样本经提取、酶解、净化、浓缩等前处理步骤之后,由UHPLC-HRMS进样分析。蜂毒肽酶解之后的特征性肽段VLTTGLPALISWIK为仪器检测的靶向物质,其精确质量数为m/z756.463 43([M+2H]2+), 其允许偏差应在5×10-6以内。该肽段的MS/MS图谱(子离子图谱)中含有丰度较高的碎片离子m/z927.564 09、585.359 38,两者的精确质量数误差应当<10×10-6。用于定量的稳定同位素内标肽段VLTTGLPALISWIK*的m/z761.478 35([M+2H]2+), 其允许偏差也在5×10-6以内。其MS/MS图谱(子离子图谱)中应该含有蜂毒肽的肽段VLTTGLPALISWIK的特异性碎片离子m/z927.564 09、585.359 38;相应的,稳定同位素内标肽段的特征肽段VLTTGLPALISWIK*的子离子图谱(MS/MS)中应含有碎片离子m/z935.604 06和m/z593.436 32,且其精确质量数的误差应当<10×10-6(见图1)。蜂毒样本中只有在精确m/z值和特征碎片离子方面同时满足以上的特征,方可肯定所求的该蜂毒样本中的VLTTGLPALISWIK含量可信,并可根据此含量的高低鉴别蜂毒质量;而且在该肽段的色谱峰附近无显著干扰峰,在定量限时,具有较好的信噪比;此外,在空白样本中,未观察到对应的信号峰,表明本研究选择的肽段具有较好的选择性与特异性。

图1 未经酶切时蜂毒肽色谱图(A);经酶切后蜂毒肽色谱图(B)

图2 未经酶切的蜂毒肽母离子质谱图(A);未经酶切的蜂毒肽二级质谱图(B);经酶切的蜂毒肽母离子质谱图(C);经酶切的蜂毒肽二级质谱图(D);未酶切时加入同位素内标质谱图(E);经酶切后加入同位素内标质谱图(F)
2.线性关系。由试验结果可知,蜂毒肽特征肽段在3~500 ng/mL的浓度范围内,线性相关系数>0.99,可见其在上述浓度范围内线性关系良好。
3.检测限、定量限。按照上文方法进行处理,结果显示,检测限为1μg/kg,定量限为3μg/kg。对蜂毒样品未酶切和经酶切后均进行检测,其检测限(LOD)、定量限(LOQ)相差较大,样品酶切后检测限、定量限更低,检测更为准确,方法学验证结果见表2和表3。
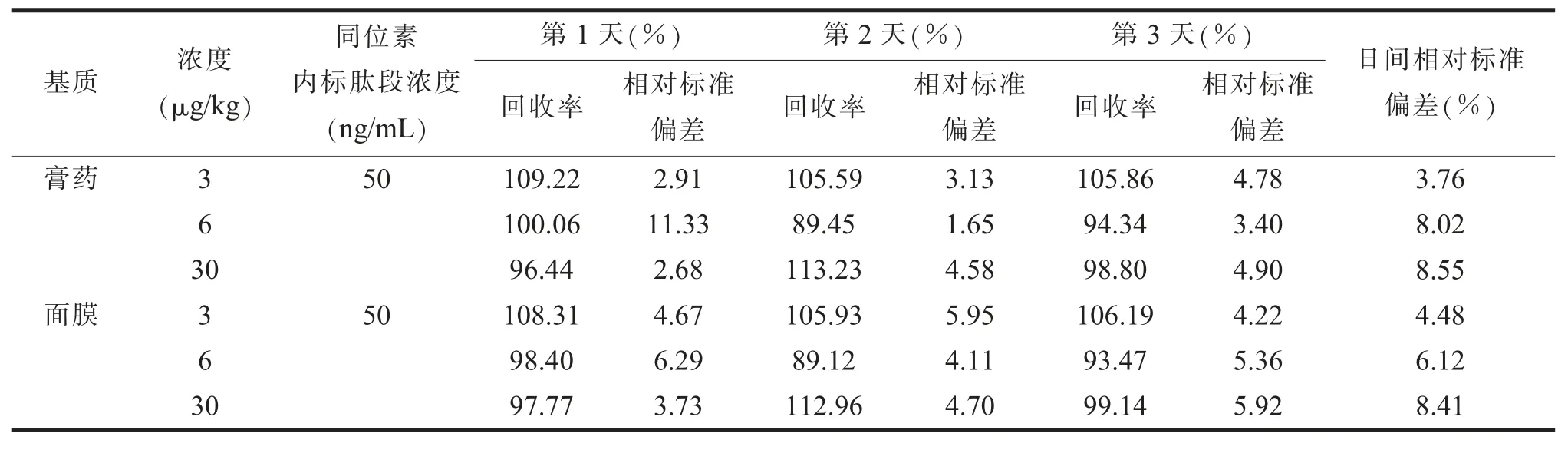
表2 方法验证相关系数

表3 未酶切与经酶切的蜂毒样品检测方法学验证结果比较
4.准确性、精密度。本试验选择特征肽段高(10LOQ)、 中(2LOQ)、 低(LOQ)3个浓度水平进行回收率和精密度试验,按上文方法前处理,进行加标回收率试验。回收率和相对标准偏差结果见表2。结果显示,在低、中、高3个浓度的添加回收率范围为89.45%~113.23%,日内与日间变异系数均<11.33%。
(三)开发的方法在实际样本检测中的应用采用本方法测定了大量样品,包括面膜、膏药、牙膏等市面标有“蜂毒”字样的产品,从质谱数据中提取肽段的质荷比,计算m/z756.463 43处特征肽段和IS肽段峰面积的比值。实测样本中蜂毒肽的检出结果见表4。结果显示,仅有26.92%的蜂毒肽衍生产品中可检测出蜂毒肽。
三、讨论
(一)仪器参数优化将特征肽段及其稳定同位素肽段的氨基酸序列输入Xcalibur软件中的Isotope simulation工具栏中,酶切得到的多肽在质谱正离子模式下通常形成带双电荷的离子的稳定形式,因此设置加合方式为[M+2H]2+,模拟特征肽段及其稳定同位素肽段的母离子和子离子质荷比。

表4 实测样本中蜂毒肽含量
将特征肽段及其稳定同位素肽段溶解、稀释为1μg/mL,在正离子模式下进行全扫(Full MS/dd MS2),调整碰撞能量和碰撞气体(氮气),对Inclusion list中包含的特征肽段进行碎裂,收集二级离子碎片信息。根据Full MS/dd MS2扫描结果,观察施加给母离子的碰撞能的碰撞效果。在二级图谱中若母离子信号强度过高则需适当增加碰撞能,反之则减小,一般选择能量增加或减小5;其次观察子离子碎片分布情况,若子离子碎片分布在较小的质荷比区域,则略微降低碰撞能,反之则增加,一般将能量以1~2增加或减小。直至母离子信号较弱、子离子质荷比又不至于过小且强度较高为止。
(二)蛋白提取方法比较不同蛋白提取方法会造成总蛋白测定结果不一,进而影响酶切效果和后期的定量结果。针对这个问题,本实验参考了5种主要的蛋白质提取方法,对不同提取方法得到的蛋白溶液,使用Bradford法蛋白质定量试剂盒定量检测蛋白浓度,结果如图3所示。综合蛋白提取浓度和试验稳定性,选择超滤管浓缩法提取蛋白。

图3 5种蛋白提取方法比较
(三)方法比较当前常用的检测方法主要是通过液相色谱串联质谱的技术定量测定完整蜂毒肽,未经酶切,检测其多电荷母离子及子离子进行定量。方法检测限、定量限较高,定量限多为40 μg/kg,不适用于复杂基质中蜂毒肽含量的检测。本方法经胰蛋白酶酶切后,可将长链多肽酶切为较短的短肽,灵敏度更高,更适用于质谱检测。
使用超高效液相色谱串联高分辨质谱(UHPLC-HRMS)对蜂毒样本进行检测,未酶切样品与酶切样品采用相同的液相色谱串联质谱方法进行检测,从结果中筛选蜂毒肽的特征肽段VLTTGLPALISWIK。对于蜂毒样品的质谱数据进行提取,选择特征肽段的母离子为m/z761.478 35([M+2H]2+)的二级质谱图,然后在二级质谱图中提取子离子m/z935.604 06和m/z593.436 32。计算、比较胰蛋白酶酶切与未酶切蜂毒样品中特征肽段/稳定同位素内标肽段峰面积比,其结果差异较大。
为提高试验中定量结果的准确性,对比之前的外标法定量,本试验选择与特征肽段性质相似的稳定同位素肽段进行定量,确保定量结果更为准确、可重复性更高,对内源性多肽的质谱信号不断增强,降低了对于给定目标蛋白的特征肽数量要求。
四、结论
本文建立超高效液相色谱串联质谱法测定复杂基质中蜂毒肽含量的分析方法,并比较两种前处理方式(胰蛋白酶酶切与未酶切蜂毒)下本方法的结果差异。结果表明,经胰蛋白酶酶切、使用同位素内标肽段后响应更高,检测限、定量限更低,检测结果更为稳定,可作为蜂毒肽衍生品中蜂毒肽含量测定的方法。
