Analysis of the HNF4A isoform-regulated transcriptome identifies CCL15 as a downstream target in gastric carcinogenesis
2021-06-19ZhenNiWenquanLuQiLiChuanHanTingYuanNinaSunYongquanShi
Zhen Ni, Wenquan Lu, Qi Li, Chuan Han, Ting Yuan, Nina Sun, Yongquan Shi
1State Key Laboratory of Cancer Biology & Institute of Digestive Diseases, Xijing Hospital, Air Force Medical University of PLA,Xi’an 710032, China; 2Department of Gastroenterology, General Hospital of Western Theater Command, Chengdu 610083,China; 3Department of Gastroenterology, First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China;4Department of Endocrinology, General Hospital of Western Theater Command, Chengdu 610083, China; 5Department of Gastroenterology, 989 Hospital of the People’s Liberation Army, Luoyang 471003, China; 6Department of Gastroenterology,
First Affiliated Hospital of Xi’an Medical College, Xi’an 710038, China
ABSTRACT Objective: Hepatocyte nuclear factor 4α (HNF4A) has been demonstrated to be an oncogene in gastric cancer (GC). However, the roles of different HNF4A isoforms derived from the 2 different promoters (P1 and P2) and the underlying mechanisms remain obscure.
KEYWORDS Gastric cancer; carcinogenesis; HNF4A; CCL15; transcriptomics
Introduction
Gastric cancer (GC) remains one of the most common malignancies, and one of the most frequent causes of cancer-related deaths1. Due to a lack of specific clinical manifestations in the early stage, most GC patients are generally diagnosed at an advanced stage with an overall 5-year survival of less than 20%, especially in China2. Therefore, increasing an understanding of GC pathogenesis may identify novel strategies for more effective treatments.
Hepatocyte nuclear factor 4α (HNF4A) has been previously shown to be an oncogene in GC3-5. Mechanistically,HNF4Ais a target ofNF-κB6, and interacts with theWntpathway3and controls tumor metabolism required for GC progression through (isocitrate dehydrogenase 1)IDH15. However, the specific target genes and downstream signaling pathways ofHNF4Aremain unknown. In addition, a total 9HNF4Aisoforms (HNF4A1-9), transcribed from P1 or P2 promoters,have been shown to display distinct tissue expressions and functions7,8(Figure 1A). Although both P1- and P2-HNF4Aare present in GC tissues9, it remains unclear which splice variant is the most relevant.
In this study, we initially determined the expressions of both P1- and P2-HNF4Ausing TCGA databases and tissue microarrays. Then, we performed transcriptome-wide mapping of both P1- and P2-HNF4Atargets in GC cell lines.Unbiased analysis revealed that the cytokine-cytokine receptor interaction andPPARsignaling pathway genes were most enriched in P1- and P2-dependent manners. We also identified chemokine (c-c motif) ligand 15 (CCL15), a member of the chemokine family, as a direct target gene of P1-HNF4A,which was required for GC progression.

Figure 1 Expression and prognostic value of P1-HNF4A in gastric cancer (GC). (A) Schematic of the HNF4A gene and its isoforms (P1 andP2). The antigens recognized by isoform-specific (P1 and P2) antibodies are labeled. (B) Representative expression of P1-HNF4A in tumor and adjacent non-tumor tissues of GC patients. Scale bars = 200 μm and 50 μm. (C) H-score of P1-HNF4A in tumor and adjacent non-tumor tissues using tissue microarray and immunohistochemical analyses (*P < 0.05). (D) The prognostic value of P1-HNF4A in GC patients according to negative and positive expression (P < 0.01).
Materials and methods
Cell culture
The human GC cell lines, AGS, MKN45, BGC823, AZ521,N87, and SGC7901, and the normal gastric epithelial cell line, GES-1 (American Type Culture Collection, Manassas,VA, USA) were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C with a humidified atmosphere of 5% CO2supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek,Israel) and 1% penicillin-streptomycin solution (Thermo Fisher Scientific). All cell line identities were verified through STR DNA profiling and were negative for mycoplasma contamination.
Cell transfection
HNF4A2(NM_000457) andHNF4A8(NM_175914) overexpression lentiviruses and a negative control were constructed by GeneCopoeia (Rockville, MD, USA). HumanCCL15-shRNA lentivirus and negative controls were constructed by GenePharma Technologies (Shanghai, China)(Table 1). Logarithmic phase cells (1 × 105) were seeded in 24-well culture plates. After adherence, lentiviral vectors were added at a final concentration of ~50 multiplicity of infection (MOI) with 0.5 μg/mL polybrene for 10 h at 37 °C.Fresh medium was replaced 10 h after transfection. The cells were transferred into 25 cm2flasks after reaching 70%-80%confluence. Then, the cell culture medium was replacedevery 2-3 days with 2 μg/mL puromycin-containing RPMI 1640 medium.
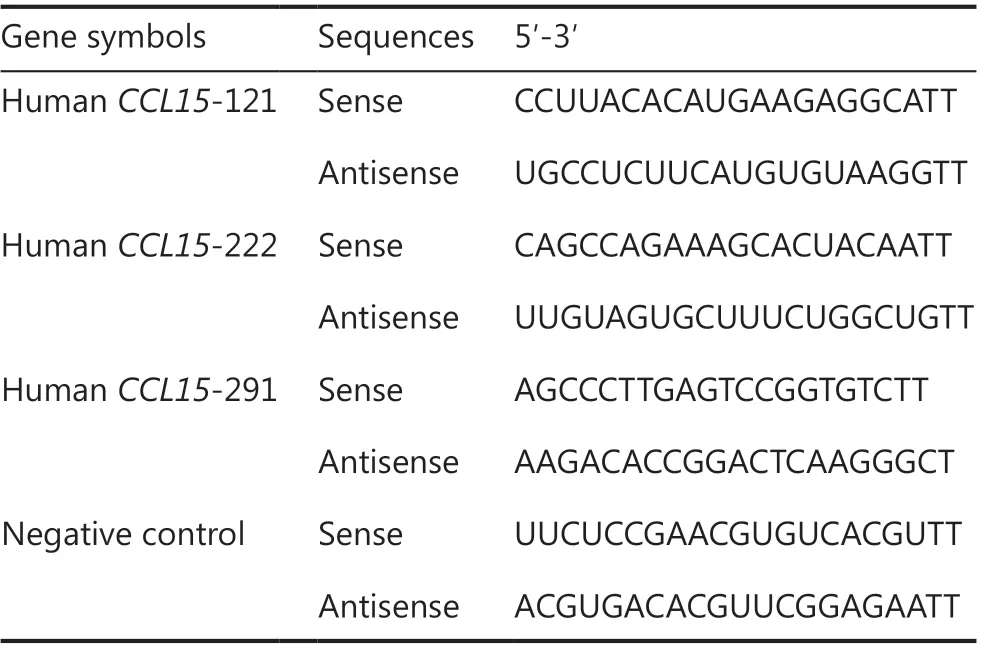
Table 1 The sequences of shRNAs used in this study
Cell proliferation assay
Cell proliferation was examined using CCK-8 kits (Thermo Fisher Scientific) according to the manufacturer’s instructions, to determine cell growth. A total of 103target cells were seeded in 96-well plates for the assays in 100 μL of complete medium. Then, 10 μL of CCK-8 reagent was added to the wells and incubated for 2 h. The cultures were assayed each day for 4 continuous days, and the absorbance was read at 450 nm with a reference wavelength at 650 nm using a Varioskan Flash Multimode Reader (Thermo Fisher Scientific). Each experiment was performed in triplicate and repeated 3 times.
Cell migration and invasion assays
Cell migration experiments were performed in a transwell chamber covered with polycarbonate membranes (Corning,NY, NY, USA). Cell invasion assays were performed using chambers coated with Matrigel matrix (BD Science, Sparks,MD, USA). Resuspended cells (1 × 105cells/well) were added to the upper chambers and cultured for 24 h. Migrated or invaded cells were stained with 0.1% Crystal Violet for 20 min at room temperature. Quantification of migrated or invaded cells was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Cell cycle assay
Target cells were seeded in 6 cm plates and harvested after reaching approximately 70%-80% confluence. Then, resuspended cells were fixed in 75% ethanol, stained with propidium iodide according to the manufacturer’s protocol (Sigma-Aldrich,St. Louis, MO, USA) and sorted using fluorescence- activated cell sorting (BD Sciences, San Jose, CA, USA). Data were analyzed using ModFit software (BD Sciences).
Murine xenograft model
Animal experiments were approved by the Institutional Animal Care and Use Committee of the Air Force Medical University of PLA (Approval No. 2018-kq-010). Briefly, 6-to 8-week-old BALB/c nude female mice (Shanghai SLAC Laboratory Animal Co., Shanghai, China) were subcutaneously implanted with 1 × 107SGC7901 cells transfected with a stable control, P1- or P2-HNF4A(n= 5). Then, the animals were euthanized and the tumor volumes were calculated using the formula (V = length × width2× 0.5) after 2 weeks.The tumor tissues were then harvested and processed for further hematoxylin and eosin staining and immunohistochemistry (IHC) analysis.
Protein lysate preparation and Western blot
Cells were digested with RIPA cell lysis buffer (Beyotime Biotechnology, Shanghai, China) with protease and phosphatase inhibitor cocktails (MCE, Shanghai, China) and were collected using a cell scraper. Total protein was extracted and quantified using the bicinchoninic acid method (Thermo Fisher Scientific). Proteins (20-30 μg) were resolved using 10% PAGE (Bio-Rad, Hercules, CA, USA), transferred to a nitrocellulose membrane (Pall Corporation, Port Washington,NY, USA) at 25 V for 30 min, and blocked for 1 h in 10%nonfat milk in 1× TBS/0.1% (v/v) Tween 20 at room temperature. Primary antibodies (GAPDH, 1:1,000, #5174;Cell Signaling Technology; β-actin, 1:1,000, #4970; Cell Signaling Technology; P1-HNF4A, 1:1,000, ab41898; Abcam;P2-HNF4A, 1:1,000, PP-H6939-00; R & D Systems; andCCL15, 1:1,000, ab219388; Abcam) were added and incubated overnight at 4 °C. Secondary antibodies (goat anti-rabbit IgG,HRP #7074 or goat anti-mouse IgG, HRP, 1:2,000, #7076; Cell Signaling Technology) were incubated at room temperature for 1 h. Signals were detected using Western Lumax Light Sirius HRP substrate reagent (ZETA-Life, San Francisco, CA,USA). All data were normalized to human β-actin orGAPDH.The bands were scanned using a ChemiDocXRS + Imaging System (Bio-Rad).
RNA-seq
Total RNA was extracted using the mirVana miRNA Isolation Kit (Ambion, Austin, TX, USA). RNA integrity was evaluated using the Agilent 2100 Bioanalyzer (Agilent Technologies,Santa Clara, CA, USA). The samples with RNA integrity number (RIN) ≥ 7 were subjected to subsequent analysis. The libraries were constructed using the TruSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, CA, USA). Then,these libraries were sequenced using the Illumina HiSeq X Ten platform, and 125 bp/150 bp paired-end reads were generated.
Transcriptome sequencing and analysis were conducted by OE Biotech (Shanghai, China). Raw data were processed using Trimmomatic (Illumina). Differentially-expressed genes(DEGs) were identified using the DESeq (2012) R package(Bioconductor; http://www.bioconductor.org/). APvalue <0.05 and fold changes > 2 or fold changes < 0.5 was set as the thresholds for significant differential expression. Hierarchical cluster analysis of DEGs was performed to characterize gene expression patterns. Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of DEGs were performed using R based on the hypergeometric distribution.
Quantitative real-time PCR
Total RNA was extracted using the RNeasy Mini Kit(QIAGEN, Duesseldorf, Germany), according to the manufacturer’s instructions. In total, 0.5 μg RNA was synthesized into cDNA using the PrimeScript RT Reagent Kit (TaKaRa,Shiga, Japan) and Mir-X mRNA First-Strand Synthesis Kit (TaKaRa) in a 10 μL volume. Real-time PCR was performed on a CFX96 system using TB Green Premix Ex Taq II (TaKaRa) with 2 μL cDNA and 0.8 μL primers in a final volume of 20 μL. The final PCR conditions were as follows:predenaturation at 95 °C for 10 min, followed by 44 cycles at 95 °C denaturation for 10 s, 60 °C annealing for 20 s, and 72 °C extension for 10 s. The target mRNA gene was normalized to humanGAPDHusing the 2-ΔΔCTmethod. Primer sequences are shown in Table 2.
Tissue microarrays and immunohistochemistry
GC tissue microarrays, including 98 cases of gastric tumor and adjunct non-tumor tissues, were obtained from Outdo Biotech(HStmA180Su15; Shanghai, China). Patient characteristics are shown in Table 3. This study was approved by the Institutional Ethics Committee of Xijing Hospital of the Air Force Medical University of PLA (Approval No. KY20183093-1).
Sections were dried overnight at 60 °C. Antigen retrieval was performed by heating in 1× citrate for 2 min and then cooling to room temperature. Endogenous peroxidase activity was blocked using 3% H2O2for 10 min. Slides were incubated with mouse anti-human antibodies against Ki67(1:1,000, ab92742), P1-HNF4A(1:100, ab41898; Abcam),P2-HNF4A(1:100, PP-H6939-00; R & D Systems), andCCL15(1:200, ab219388; Abcam) at 4 °C overnight. Thesections were then counterstained with hematoxylin staining and dehydrated.

Table 2 The sequences of primers used in this study
The slides were scanned and viewed using a Pannoramic Viewer (3DHISTECH, Budapest, Hungary). The staining intensity of P1- and P2-HNF4Awas semiquantitatively determined using the H-score method10. Only unequivocally stained nuclei were considered positive. H-scores < 50 and≥ 50 were considered as negative and positive, respectively.H-scores < 150 and ≥ 150 were considered low and high expressions, respectively.CCL15staining was semiquantitatively performed according to the staining intensity and percentage of positive cells11. IHC scores < 6 and ≥ 6 were considered low and high expressions, respectively.
Dual-luciferase reporter assays
Briefly, 2,000 bp fragments of the humanCCL15promoter were obtained from Ensembl and predicted in the JASPAR database. The wild-type (WT) and corresponding mutationalCCL15promoter fragments covering predicted DR1 sites were PCR-amplified and cloned into the firefly luciferase reporter plasmid, pGL3-basic vector (Promega, Madison,WI, USA). Luciferase activity was then detected using a Dual-Luciferase Reporter Assay Kit (Promega) at 48 h after reporter transfection using Lipofectamine 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) in SGC7901 cells.Firefly luciferase activity was normalized to Renilla luciferase activity, and the final data are presented as the fold induction of luciferase activity compared to that of the negative control.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed using the EZ ChIP™ Kit(Millipore, Billerica, MA, USA). The cells were cross-linked with 1% formaldehyde for 10 min at 37 °C and quenched with 2.5 M glycine for 5 min at room temperature. DNA was immunoprecipitated from the sonicated cell lysates usingHNF4Aantibody (Abcam, Cambridge, MA, USA) and subjected to PCR to amplify the HNF4 binding site (Table 2). The amplified fragments were analyzed on an agarose gel. A nonspecific antibody against IgG served as a negative control.
Data mining using public databases
The mRNA expression and DNA copy number ofHNF4Ain GC were analyzed within the Oncomine (http://oncomine.org), GEPIA (http://gepia.cancer-pku.cn), and Ualcan (http://ualcan.path.uab.edu) databases. The GEPIA and Ualcan databases were used to analyze candidate gene alterations that were identified as being enriched in RNA-seq. We further used the LinkedOmics (http://www.linkedomics.org), cBioPortal(http://cbioportal.org), and GEPIA databases to characterize the relationships betweenHNF4Aand candidate target genes.
Statistical analysis
All cell culture experiments were performed in triplicate to reduce error and ensure reproducibility. All quantitative data are expressed as the mean ± SEM. Differences between two groups were examined using an unpaired Student’st-test.Differences between multiple groups were compared by oneway analysis of variance with Dunnett’s post hoc tests. All categorical data are expressed as rates. Differences between two groups were examined using the χ2test. Statistical analysis was performed using SPSS 13.0 statistical software for Windows(SPSS, Chicago, IL, USA) or GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).P< 0.05 was considered statistically significant.
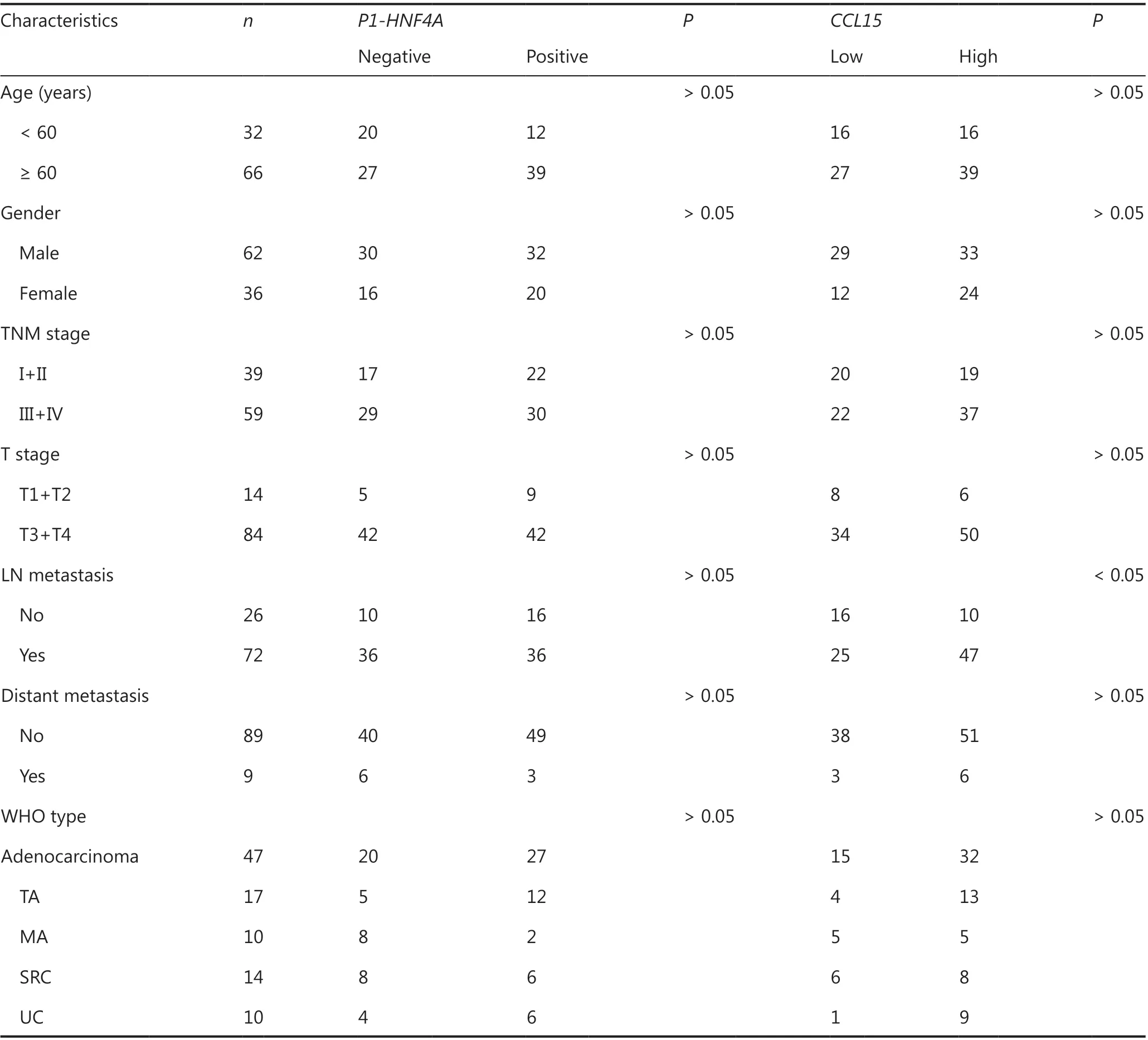
Table 3 Clinical characteristics of the patients analyzed using tissue microarrays
Results
The expression levels of HNF4A in TCGA databases
Initially, the mRNA expression and DNA copy number ofHNF4Ain GC were analyzed within the Oncomine, GEPIA,and Ualcan databases. Data in the Oncomine database revealed that mRNA expression and DNA copy number variation ofHNF4Awere significantly higher in GC tissues than in normal tissues in TCGA Gastric, Deng Gastric, Cho Gastric, and DErrico Gastric (P< 0.01), especially for intestinal type adenocarcinomas (P< 0.01) (Supplementary Figure S1). Data in the Ualcan database also showed significantly higher expression levels ofHNF4Ain tumor tissues than in non-tumor tissues (Supplementary Figure S2A).Early stages showed significantly higherHNF4Aexpression levels than those in late stages (Supplementary Figure S2B and S2C). Finally, data in the GEPIA database also revealed significantly higherHNF4Aexpression levels in tumor tissues than in non-tumor tissues (Supplementary Figure S3A).Furthermore, isoform analysis indicated thatHNF4A1andHNF4A2expressions were significantly higher thanHNF4A7andHNF4A8expressions (Supplementary Figure S3B).Together, these results showed thatHNF4Aamplification was common in GC, especially in the intestinal type and early stages. In addition, P1-HNF4Aupregulation was more remarkable than P2-HNF4Aupregulation.
The expression levels of P1- and P2-HNF4A in gastric tumors and adjacent tissues
Although highHNF4Aexpression has been shown in GC tissues3-5, previous studies did not distinguish the functions of differentHNF4Aisoforms. We therefore detected the expression levels of both P1- and P2-HNF4Ain gastric tumors and adjacent non-tumor tissues using tumor microarrays. The results indicated that P1-HNF4Aexpression was significantly increased in tumor tissues compared with non-tumor tissues (P< 0.05) (Figure 1B and 1C). However, no significant difference was noted in P2-HNF4Aexpression (P> 0.05)(Supplementary Figure S4A and S4B), although the H-score of P2-HNF4Awas higher in non-tumor tissues than in tumor tissues (P> 0.05) (Supplementary Figure S4B). Next, we examined P1-HNF4Aexpression according to cancer clinical features. As shown in Table 2, P1-HNF4Aexpression levels showed an upregulation in the I+II, T1+T2, M0, and N0 stages, but no significant difference was noted (P> 0.05).Finally, patients with positive P1-HNF4Astaining showed a significantly shorter survival compared with negative cases(P< 0.05) (Figure 1D). However, no significant difference was noted between low and high P2-HNF4Aexpression patients(Supplementary Figure S4C). Together, these results indicated that P1- and P2-HNF4Amight play different roles during GC progression. P1-HNF4Aexpression was the main isoform in gastric carcinogenesis and predicted poor prognoses.However, no significant difference was observed in P2-HNF4Aexpression levels.
P1- and P2-HNF4A showed distinct functions both in vitro and in vivo
We further examined the roles of both P1- and P2-HNF4Ain GC cellsin vitroandin vivo. First, we detected the expression levels of P1- and P2-HNF4Ain GC cell lines. Western blot results indicated that P1- and P2-HNF4Aexpression levels showed dramatic differences (Supplementary Figure S5A).Then, we directly introduced P1-HNF4A(HNF4A2) and P2-HNF4A(HNF4A8) into SGC7901 and BGC823 cells,respectively, 2 cell lines with relatively low P1- and P2-HNF4Aexpressions (Supplementary Figure S5B). The CCK-8 assay indicated that P1-HNF4Aoverexpression promoted GC cell proliferation (P< 0.01) (Figure 2A). However, no significant difference was noted after P2-HNF4Aoverexpression (P> 0.05) (Figure 2A). Furthermore, flow cytometry analysis showed that P1-HNF4Aoverexpression promoted G1/S phase arrest (P< 0.01), and P2-HNF4Aoverexpression showed no significant effect on the cell cycle (P> 0.05)(Figure 2B). Furthermore, P1- and P2-HNF4ApromotedERK1/2pathway activation, with a more significant tendency in P1-HNF4A-overexpressing cells (Supplementary Figure S5B).
Next, transwell invasion and migration assays also showed that P1-HNF4Aoverexpression facilitated GC cell invasion and migration (P< 0.01), but P2-HNF4Aoverexpression showed no effect (P> 0.05) (Figure 2C and 2D). Moreover,we performed anin vivoxenograft mouse experiment using stably transfected SGC7901 cells. The results showed that the average tumor volume in the P1-HNF4Agroup was significantly larger than that in the control group (P< 0.05) (Figure 3A and 3B). However, no significant difference was found in the average tumor volume in the P2-HNF4Agroup compared with the control (P> 0.05) (Figure 3A and 3B). In addition,tumors from P1-HNF4Aoverexpression mice showed more significantKi67expression than those from P2-HNF4Aoverexpression mice (Figure 3C and 3D).
Taken together, the abovein vitroandin vivoresults indicated that P1-HNF4Acould significantly promote GC progression, and that P2-HNF4Ashowed no significant effect.
RNA-seq showed distinct pathway and gene enrichment in P1- and P2-HNF4Aoverexpressing cells
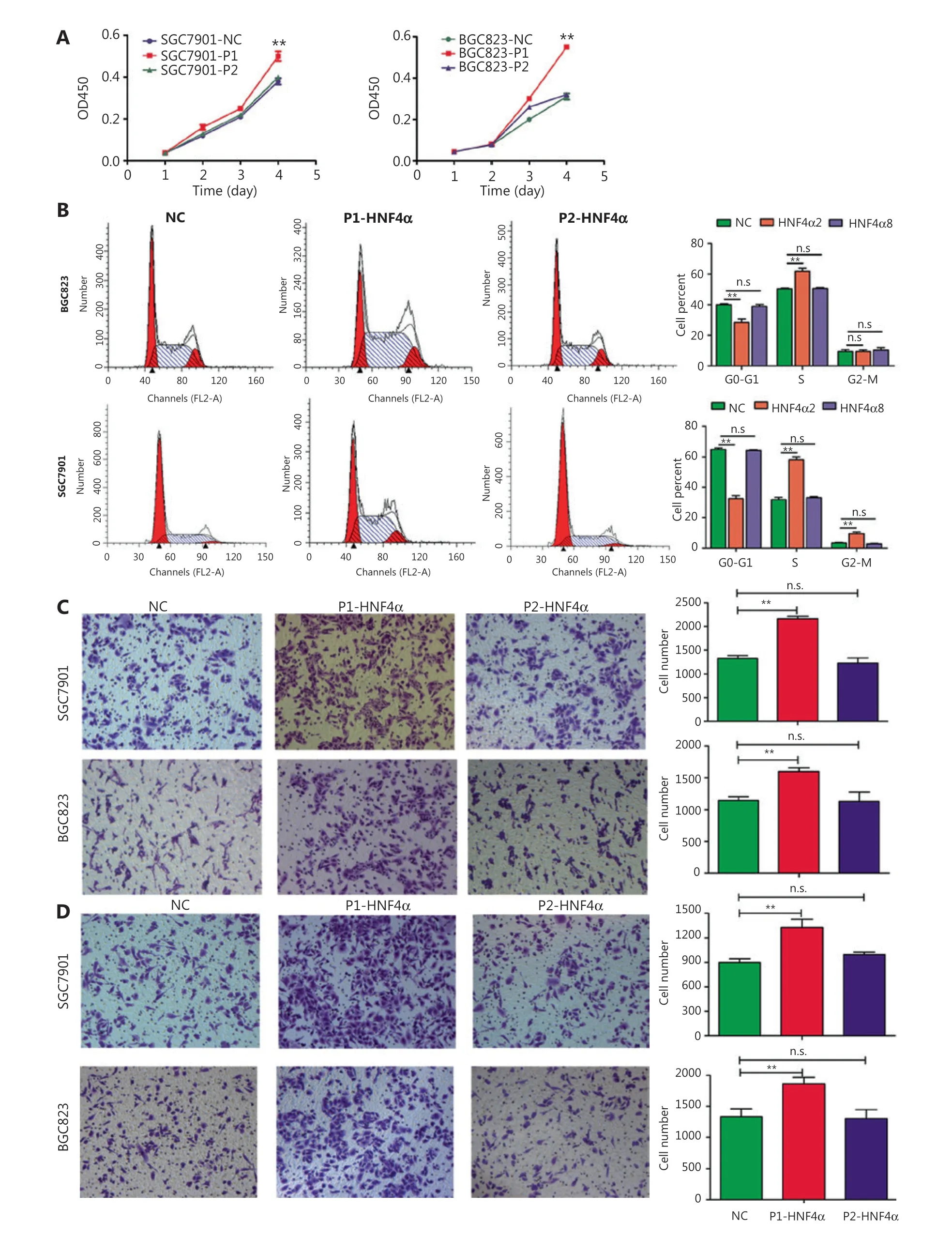
Figure 2 The effects of both P1- and P2-HNF4A on malignant phenotypes of gastric cancer cells in vitro. (A) P1-HNF4A or P2-HNF4A overexpression lentivirus were transfected into SGC7901 and BGC823 cells. Cell proliferation was examined using the CCK8 assay. OD450 absorbance was detected every day for a continuous 4 days (**P < 0.01, n.s., nonsignificant, n = 3). (B) The effects of P1- and P2-HNF4A overexpression on SGC7901 and BGC823 cell cycles using a flow cytometry assay (**P < 0.01; n.s., nonsignificant, n = 3). (C) The effects of both P1- and P2-HNF4A overexpression on SGC7901 and BGC823 cell migrations using transwell experiments. Migrated cells were counted using ImageJ (**P < 0.01;n.s., nonsignificant, n = 3) (Crystal Violet staining, 20×). (D) The effects of both P1- and P2-HNF4A overexpression on SGC7901 and BGC823 cell invasion using transwell experiments. Invaded cells were counted by ImageJ (**P < 0.01; n.s., nonsignificant, n = 3).

Figure 3 The roles of both P1- and P2-HNF4A on gastric cancer (GC) progression in vivo using murine xenograft experiments.(A) BALB/c nude mice underwent orthotopic implantation with SGC7901 cells stably transfected by a negative control, P1-HNF4A, and P2-HNF4A overexpression lentiviruses. (B) The tumors were extracted and measured. Tumor volume was calculated using the formula(V = length × width2 × 0.5). The results are expressed as the mean ± SEM (*P < 0.05, n.s., nonsignificant). (C) H&E staining of sections from orthotopic specimens. Scar bars = 200 μm and 50 μm). (D) Ki67 staining of section from orthotopic specimens by immunohistochemistry.Scale bars = 200 μm and 50 μm).
To investigate the underlying mechanisms mediating P1-and P2-HNF4Afunctions, we performed RNA-seq using P1- and P2-HNF4A-overexpressing SGC7901 cells. For this purpose, we transfected SGC7901 cells with P1-HNF4A,P2-HNF4Aand negative control lentiviruses. Increased expression of P1- and P2-HNF4Aupon transfection was confirmed by Western blot (Supplementary Figure S5B).Then, we performed RNA-seq to examine the effects of P1- and P2-HNF4Aon the SGC7901 transcriptome.We found that both P1- and P2-HNF4Aoverexpression resulted in considerable changes in gene expression at the mRNA level (Supplementary Figure S6A). Using 2-fold as a cutoff to designate DEGs, we found 310 and 105 genes that were downregulated and 763 and 190 genes that were upregulated in P1- and P2-HNF4A-overexpressing cells,respectively (Supplementary Figure S6B). There was an overall greater effect in P1-HNF4Athan in P2-HNF4Ain terms of the number of dysregulated genes with large fold changes, which could be because P1-HNF4Atypically has a more potent transactivation function than P2-HNF4A.KEGG pathway analysis of the differentially regulated genes showed that there was a significant upregulation of genes involved in the cytokine-cytokine receptor interaction, and a decrease in genes involved in the calcium signaling pathway in P1-HNF4Acells (Figure 4A). In P2-HNF4Acells,there was a significant upregulation of genes involved in thePPARsignaling pathway and a decrease in genes involved in valine, leucine, and isoleucine biosynthesis (Figure 4B).
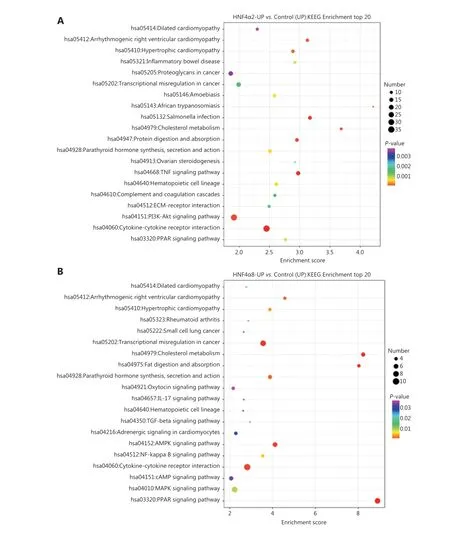
Figure 4 RNA-seq screen of target genes and signaling pathways using downstream P1- and P2-HNF4A overexpression in SGC7901 cells.(A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showing enrichment of the top 20 signaling pathways in P1-HNF4A overexpression compared to the negative control in SGC7901 cells. (B) KEGG pathway analysis showing enrichment of the top 20 signaling pathways in P2-HNF4A overexpression compared to the negative control in SGC7901 cells.
CCL15 is a direct target of P1-HNF4A and is functionally required in GC
To test the deregulated genes involved in the cytokine-cytokine receptor interaction pathway in GC development, we examined the expression of these genes within TCGA databases. Data in GEPIA revealed that the expressions ofCCL5,CCL15,CCL20,CSF2RA,CXCL1,CXCL8,CXCL16,CXCR4,GDF15,IL1RN,IL2RB,IL2RG,IL22RA1,INHBA,TNFRSF1B,andTNFRSF14were significantly higher in GC tissues than in normal tissues (Supplementary Figure S7). Data from Ualcan revealed that the expressions ofBMP2,CCL5,CCL15,CCL20,IL8,CD70,CSF2RA,CXCL1,CXCL16,CXCR4,TGFB2,GDF15,IL11,IL1R1,IL1RN,IL13RA2,IL32,IL2RB,IL2RG,IL22RA1,IL18R1,IL18RAP,INHBA,INHBB,INHBE,TNFRSF9,TNFRSF1B, andTNFRSF14were significantly higher in GC tissues than in normal tissues (Supplementary Figure S8).
Among the aforementioned gene deregulations in GC, we focused on chemokines, includingCCL15, the role of which remained unclear in GC progression. Then, we examined thein vitrorole ofCCL15on GC cell proliferation and invasion.Initially, we detected the mRNA expression ofCCL15in the normal gastric epithelial cell line, GES-1, and the GC cell lines,BGC823, MKN45, AZ521, SGC7901, and AGS. Notably, the mRNA level ofCCL15in GC cells far surpassed that in GES-1 cells (Supplementary Figure S9). Then, shRNA-mediatedCCL15knockdown cells were generated in MKN45, a cell line with relatively highCCL15expression (Figure 5A). Significant reductions in the proliferation rate were observed in shCCL15cells compared with control cells using the CCK-8 assay (P<0.01) (Figure 5B). Significantly reduced cell migration and invasion were also observed in shCCL15cells compared with control cells in the MKN45 cell line (P< 0.01) (Figure 5C and 5D). To determine whetherCCL15was also upregulated in primary GCs, we analyzedCCL15expression using a GC tissue microarray. The results indicated thatCCL15expression was significantly higher in tumors than in paracancerous tissues (P< 0.01) (Supplementary Figure S10A and S10B).Subgroup analysis indicated that patients with highCCL15expression showed more lymph node metastasis than those with lowCCL15expression (P< 0.05) (Table 3). Survival analysis showed that patients with highCCL15expression had a poorer prognosis than patients with lowCCL15expression(P< 0.01) (Figure 5E). We further explored the prognostic value ofCCL15in the cBioportal database. HighCCL15expression showed poorer prognosis in both the Firehose legacy and PanCancer Atlas groups (Figure 5F and 5G). In summary, these results supportedCCL15as an oncogene in GC progression.
To check ifCCL15acted as a downstream target, we performed qRT-PCR. The results indicated that both P1- and P2-HNF4Acould significantly upregulateCCL15expression,with P1-HNF4Abeing more prominent (P< 0.01) (Figure 6A). Then, we examined the promoters ofCCL15using JASPAR. The results showed that the promoter ofCCL15contained two predicted DR1 motifs (AGGTCAnAGGTCA)(Figure 6B). Next, we constructed luciferase reporter gene fragments covering the two DR1 sequences (wild-type and mutant) (Figure 6B). The results revealed thatHNF4A2positively regulated theCCL15promoter containing the two DR1 sites between ~81~95 and ~243~257 (Figure 6B and 6C).Chromatin immunoprecipitation (ChIP) assays further confirmed thatHNF4A2bound to the speculative sites (~81~95 and ~243~257) on theCCL15promoter in SGC7901 cells(Figure 6D). Furthermore, to determine the clinical relevance,we divided the GCs into P1-HNF4A-negative and P1-HNF4Apositive subgroups according to the H-score.CCL15high expression cases in the P1-HNF4A-positive group were found to be significantly higher than those in the P1-HNF4A-negative group (P< 0.01) (Figure 6E and 6F). We further determined the correlation betweenHNF4AandCCL15in TCGA databases. Both data in the GEPIA, cBioportal, and LinkedOmic databases revealed that HNF4A was positively correlated withCCL15(Figure 6G, 6H, and 6I).
Discussion
Accumulating evidence has revealed the vital role ofHNF4Ain GC initiation and progression. However, the functions of differentHNF4Aisoforms and underlying molecular mechanisms in GC cell growth and invasion have not been completely clarified. In this study, we demonstrated that P1-HNF4Awas the main isoform upregulated in GC progression. Furthermore,we revealed that the cytokine-receptor pathway was one of the key downstream signaling pathways. We also demonstrated thatCCL15was the most abundant chemokine downstream of P1-HNF4Ain human GC, with significant prognostic value.These findings revealed complex tumor-promoting effects shaped by theHNF4A-CCL15axis in human GC.
HNF4Ais a highly conserved member of the nuclear receptor superfamily, and regulates genes involved in hepatocyte differentiation12,13. It also plays key roles in kidney14, pancreas15, and gut16development, and regulates the expression of genes involved in metabolism17,18, homeostasis19-21, differentiation22, and immune response23,24. The roles and functions ofHNF4Ain HCC and CRC have been clearly defined25-27.In contrast, the role ofHNF4Ain stomach development and GC has only preliminarily been investigated. Recent studies showed thatHNF4Ais functionally required for GC survival by interacting with theWntandNF-κBpathways and regulating tricarboxylic acid (TCA) cycle metabolism3,5,6. In contrast,another recent study showed that the tumor suppressorITLN1promotedHNF4Aexpression by repressingNF-κBin GC28,and high HNF4A expression showed improved survival outcomes28. Similar inconsistent results have also been observed in CRC27,29. These paradoxical conclusions indicated that the functions ofHNF4Awere characterized with time and spatial distribution characteristics. The expression ofHNF4Amight be dynamically regulated during GC initiation and progression. Our preliminary results found thatHNF4Awas upregulated in GC, especially in the early stage, which supported the oncogenic roles ofHNF4A.
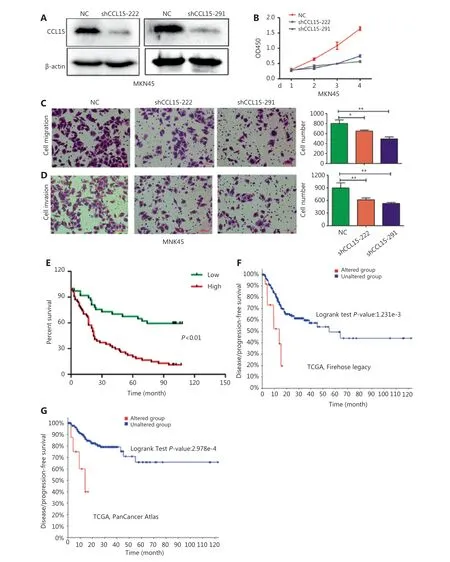
Figure 5 The effects of CCL15 on malignant phenotypes of gastric cancer (GC) cells in vitro. (A) Negative control and sh-CCL15 lentivirus were transfected in MKN45 cells and CCL15 protein expression was detected by Western blot. β-actin was used as internal control. (B) MKN45 cell viability infected with lentivirus expressing shRNAs targeting CCL15 or control shRNA was examined by CCK8, and OD450 was detected at the indicated time (**P < 0.01, n = 3). (C, D) Cell migration and invasion was performed using transwell assays. Migrated (C) or invaded (D)cells were counted by ImageJ (*P < 0.05, **P < 0.01, n = 3) (Crystal Violet staining, 20×). (E) The prognostic value of CCL15 in GC patients using tissue microarrays. (F) The prognostic value of CCL15 in GC patients of the Firehose legacy cohort using the cBioPortal database (Z-Score = 2).(G) The prognostic value of CCL15 in GC patients of the PancancerAtlas cohort using the cBioPortal database (Z-Score = 2).
A total of nineHNF4Asplice variants have been generated via two alternative promoters (P1 and P2 promoters) with tissue- and cell-specific expression patterns30,31. P1-drivenHNF4AincludesHNF4A1-6isoforms and P2-drivenHNF4AincludesHNF4A7-9isoforms respectively30. P1-drivenHNF4Ais expressed in the adult liver32and kidney7, whereas P2-drivenHNF4Ais expressed in the fetal liver, the adult stomach7and pancreas30. Both P1- and P2-HNF4Aare expressed in the adult colon and intestine, but with distinct location distributions30. The unique expression patterns of P1- and P2-drivenHNF4Asuggested distinct functional roles of differentHNF4Aisoforms under physiological conditions. Furthermore, previous research has demonstrated thatHNF4Aisoforms showed significant metabolic differences and varied susceptibilities to colitis-associated colon cancer in exon-swap mice33,34. In gastric carcinogenesis, both P1-and P2-HNF4Awere positive in gastric precancerous lesions and tumor tissues9,35, which suggests that theHNF4Agene may exhibit oncogenic activity in GC. However, the relative contributions of the differentHNF4Aisoforms (P1- and P2-HNF4A) have not been determined.
P1-HNF4Aacts as a tumor suppressor both in HCC and CRC, inhibiting cell proliferation and the inflammation pathway25,34. In contrast, P2-HNF4Apromotes colitis and colitis-associated colon cancer development34. Previous studies showed that stomach tissues were positive for only P2-HNF4A7.In contrast, Moore et al.19indicated thatHNF4A, especially P1-HNF4A, regulated gastric epithelium homeostasis and was necessary for the maintenance of ZC secretory architecture. In this study, we demonstrated that P1- and P2-HNF4Aexhibited significantly different functions during GC progression.However, unlike in HCC36and CRC37, P1-HNF4Awas the main oncogene, while P2-HNF4Aexpression was irrelevant to GC. Previous studies have demonstrated that P1-HNF4Aplays key roles in both intestinal epithelium and liver cell development and differentiation16,23, so it is possible that tissue specific transcription factors might suppress tumorigenesis. In contrast, ectopic expression of lineage-survival oncogenes may promote cancer by reactivating early developmental programs. These results were consistent with the high expression of P1-HNF4Ain intestinal-type GC4, so we proposed that the function ofHNF4Awas tissue-specific, which might account for the distinct roles of P1- and P2-HNF4Ain different cancer development. Furthermore, integration of RNA-seq data suggests that this functional difference is due to differential expression of certain target genes, withHNF4A2upregulating genes involved in cell proliferation and inflammation, andHNF4A8upregulating genes involved in metabolism. In addition, P1-HNF4Atypically has a more potent transactivation function than P2-HNF4A, which displays a similar pattern as in CRC.
Recent evidence suggests the involvement ofHNF4Ain the pathophysiology of inflammatory diseases, such as inflammatory bowel diseases24,38,39.HNF4Ahas also been shown to interact with theIL-6/STAT3andNF-κBpathways6,25. Additionally,HNF4Awas involved inIL-1β signal transduction through the regulation ofIL-1R6. These results indicated that inflammation regulation might be a potential molecular mechanism involving the functional roles ofHNF4A. In this study, we identifiedCCL15as a direct P1-HNF4Atarget gene that promoted GC cell proliferation and invasion.CCL15has been shown to play a critical role in tumor progression in various cancer types, including liver and colorectal cancers40-42. However, the role ofCCL15in GC remains unclear. Accumulating evidence has suggested thatCCL15may have a crucial role in the progression of tumor cells via the CC chemokine receptor42,43. Previous studies have preliminarily indicated thatCCL15expression is higher in gastric tumor tissues than in normal tissues44,45.In human CRC cells, loss ofSMAD4leads to the upregulation ofCCL15expression40,42,43. Additionally, in human liver cancer cells, both inflammatory factors and epigenetics regulateCCL15expression41. Mechanistically,CCL15could promote tumor progression through the only receptor,CCR1, which is expressed by both immune cells such as monocytes41, lymphocytes46, neutrophils40, eosinophils47and tumor cells41,48. Our results revealed thatCCL15expression was higher in GC tissues than in normal tissues, and predicted a poor prognosis.CCL15knockdown significantly repressed GC cell proliferation, which indicated thatCCL15might promote GC cell proliferation in an autocrine manner (Supplementary Figure S11). WhetherCCL15promotes GC progression through recruitment ofCCR1immune cells in a paracrine manner requires further exploration. More importantly, our results indicated the positive correlation of P1-HNF4AwithCCL15in a subset of GC patients. Thus,aberrant P1-HNF4Aexpression in certain GC patients might provide a possible clue for the development of an effective therapy byCCL15signaling pathway blockade.
Conclusions
In summary, our study preliminarily showed the biological and clinical significance of differentHNF4Aisoforms in GC. Compared to P2-HNF4A, P1-HNF4Amight be the main driver oncogene in GC progression. Mechanistically,P1-HNF4Adirectly regulated the cytokine-receptor pathway,thereby promoting tumor growth and progression.CCL15could be regarded as a biomarker to guide therapy in GC patients with a high expression of P1-HNF4A, and pharmaceutical intervention of theCCL15signaling pathway may provide a promising strategy to improve the prognoses of GC patients.
Grant support
This work was supported by the National Natural Science Foundation of China (Grant No. 81873554) and Shaanxi Foundation for Innovation Team of Science and Technology(Grant No. 2018TD-003).
Conflict of interest statement
No potential conflicts of interest are disclosed.
Availability of data and materials
The RNA-seq datasets generated and analyzed during the current study are available in the NCBI SRA repository(PRJNA612930).
杂志排行

Cancer Biology & Medicine的其它文章
- Circular RNAs: new biomarkers of chemoresistance in cancer
- Biological roles and potential clinical values of circular RNAs in gastrointestinal malignancies
- Biomaterial-based platforms for cancer stem cell enrichment and study
- sLex expression in invasive micropapillary breast carcinoma is associated with poor prognosis and can be combined with MUC1/EMA as a supplementary diagnostic indicator
- Nanomaterial-based delivery vehicles for therapeutic cancer vaccine development
- Targeting FGFR in non-small cell lung cancer: implications from the landscape of clinically actionable aberrations of FGFR kinases