基于16s rRNA的徽派腊肉加工过程中微生物群落结构分析
2021-06-15周莹王兆明涂健陈兴勇徐宝才
周莹 王兆明 涂健 陈兴勇 徐宝才


摘要:为探究徽派腊肉加工过程中的微生物群落演替规律,采用高通量测序技术对不同加工阶段的徽派腊肉中微生物16S rRNA进行V3~V4区测序,比较不同加工阶段(鲜肉、腌制中期、腌制结束、成熟中期、成熟结束)腊肉中细菌群落结构组成及多样性差异。结果表明:5 个加工阶段分别获得80 155、80 116、80 174、80 114、80 174 条有效序列;在门分类水平上,厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinomycetes)、拟杆菌门(Bacteroides)相对丰度较高;在属分类水平上,鲜肉、腌制中期和腌制结束的腊肉中主要优势菌为乳球菌(Lactococcus)和假單胞菌(Pseudomonas),成熟中期和成熟结束的腊肉中主要优势菌为葡萄球菌(Staphylococcus)、盐弧菌(Salinivibrio)和放线菌(Actinomycetes)。微生物群落结构和多样性在徽派腊肉不同加工阶段存在明显差异。
关键词:徽派腊肉;高通量测序;发酵;微生物群落;细菌多样性
Analysis of Microbial Community Structure during the Processing of Hui-Style Bacon by 16S rRNA Sequencing
ZHOU Ying1,2,3, WANG Zhaoming2,3, TU Jian1, CHEN Xingyong1, XU Baocai2,3,*
(1.School of Tea and Food Science and Technology, Anhui Agricultural University, Hefei 230036, China; 2.School of Food and Biological Engineering, Hefei University of Technology, Hefei 230009, China; 3.Engineering Research Center of Bio-process, Ministry of Education, Hefei University of Technology, Hefei 230009, China)
Abstract:High throughput sequencing (HTS) was used to investigate the microbial community succession of Hui-style bacon samples at different processing stages (fresh meat, mid-curing, the end of curing, mid-maturation and the end of maturation). The V3-V4 region of the bacterial 16S rRNA gene was sequenced to compare the composition and diversity of bacterial community among the processing stages. The results showed that 80155, 80116, 80174, 80114 and 80174 effective sequences were obtained for the five processing stages, respectively. At the phylum level, Firmicutes, Proteobacteria, actinomycetes and Bacteroides were dominant. At the genus level,LactococcusandPseudomonaswere the dominant bacteria in fresh meat, at the mid-cuing stage and at the end of curing, whileStaphylococcus,Salinivibrioandactinomyceteswere the dominant bacteria at the mid-maturation stage and at the end of maturation. These findings showed that different processing stages had significant effects on the microbial community structure and diversity in Hui-style bacon.
Keywords:Hui-style bacon; high throughput sequencing; fermentation; microbial community; bacterial diversity
DOI:10.7506/rlyj1001-8123-20201215-289
中图分类号:TS251.5 文献标志码:A 文章编号:
引文格式:
周莹, 王兆明, 涂健, 等. 基于16s rRNA的徽派腊肉加工过程中微生物群落结构分析[J]. 肉类研究, 2021, 35(2): . DOI:10.7506/rlyj1001-8123-20201215-289. http://www.rlyj.net.cn
ZHOU Ying, WANG Zhaoming, TU Jian, et al. Analysis of microbial community structure during the processing of hui-style bacon by 16S rRNA sequencing[J]. Meat Research, 2021, 35(2): . DOI:10.7506/rlyj1001-8123-20201215-289. http://www.rlyj.net.cn
腌腊肉制品在我国有上千年的制作历史[1-2],深受人民群众的喜爱。生肉经过腌制和成熟后,肉色腊红、耐贮藏、色味鲜美、食用方便,有着独特的腌腊风味[3-4]。徽派腊肉俗名“刀板香”,选料精良,制作复杂,原料采用徽州地区土杂猪厚度均匀的五花肉,在适当的环境条件下(温度、湿度),经预处理、腌制、晾晒(轻发酵)等核心工艺加工而成,是具有典型特征品质的腌腊肉制品。徽派腊肉成品具有高盐含量和低水分含量的特点,兼具干腌肉制品和发酵肉制品特性。作为徽菜的典型代表,徽派腊肉是徽州文化的一张重要名片,是具有浓郁地方特色的中式传统肉制品。
微生物的代谢产物与酶会对肌肉蛋白的降解、脂肪的氧化水解等有重要影响,在发酵过程中对腊肉风味轮廓形成具有重要贡献[5]。有研究表明,腌腊肉制品的质量与微生物的活动密切相关[6-7]。Yu Hai等[8]从如皋火腿中分离出1 株高效的转氨酶生产菌(肺炎克雷伯菌YZusK-4),并成功将其应用到发酵肉制品中,达到显著改善产品风味的效果。Zhao Changqing等[9]研究发现,嗜酸乳杆菌作为发酵剂,不仅可以改善肉品风味,还可以有效抑制有害微生物生长,提高食用安全性。Berdague等[10]通过在发酵香肠中接种发酵型乳酸菌和葡萄球菌等,发现可以产生78 种风味物质,如烃、醛、酮等。类似地,Bruna等[11]通过在发酵香肠中接种霉菌,发现接种后香肠的保质期显著延长,同时香肠的风味物质种类也显著增加。不难发现,微生物在发酵肉制品风味形成中起重要作用。全面了解腊肉的微生物群落演替有助于解析其风味形成机制,并且可以为天然发酵剂的制备提供一定的理论依据。
高通量测序(high-throughput sequencing,HTS)是食品行业的一项新兴技术,可以准确识别不同食物基质中的微生物群落,并且能够快速、准确地获取丰富的生物学数据和大量微生物学信息,已成为研究微生物多样性和群落特性的主要方法之一[12-13]。HTS不仅可以提供有关微生物群落结构的信息,还可以提供实用的基因组成和相关分布[14]。HTS技术在许多发酵食品中得到了广泛应用,如豆豉[15]、泡菜[16]、酸菜[17]和发酵香肠[18]等。目前,HTS技术也已经被用来研究腊肉中的微生物群落结构[19-20],但对徽派腊肉微生物结构的相关报道还很少。本研究基于HTS技术揭示徽派腊肉发酵过程中微生物的变化,旨在为探明徽派腊肉的品质形成机制和商业化开发提供一定的参考。
1 材料与方法
1.1 徽派臘肉样品采集
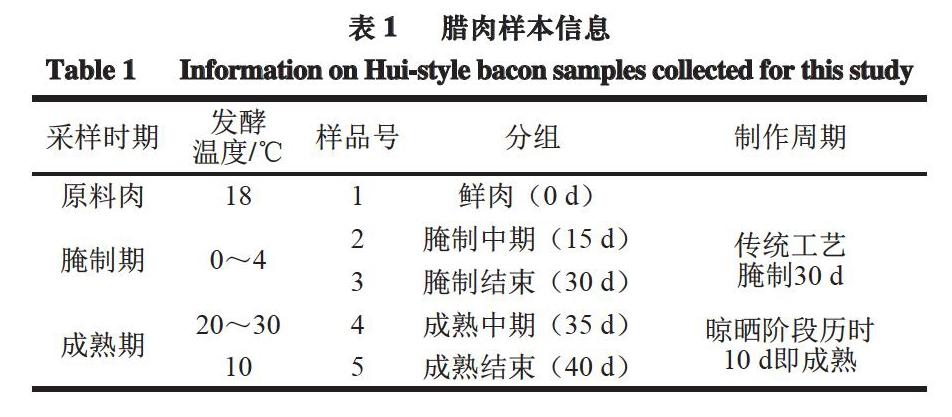
跟踪采集徽派腊肉(2019年5月—2019年7月)1 个批次(约1 t)生产的完整过程。根据生产周期确定5 个采样点(见表1)。取样完成后,当天将样品运回实验室分离,每个样品不少于1 000 g,每个时间点取3 个平行样品,混匀。准确称取10 g样品,分装至无菌真空袋中,按不同取样周期编号。以上过程均在超净微生物检测台上完成。
细菌基因组DNA快速提取试剂盒 天根生化科技(北京)有限公司;515F/806R引物 武汉天一辉远生物科技有限公司;DNeasy mericon Food Kit提取试剂盒 德国Qiagen公司;5×Trans Start?Fast Pfu缓冲液 生工生物工程(上海)股份有限公司;Phusion?High-Fidelity PCR Master Mix with GC Buffer 美国New England Biolabs公司;其他试剂均为国产分析纯。
1.2 仪器与设备
DYY-6c电泳仪 北京六一仪器厂;WD-943B凝胶成像系统 北京六一生物科技有限公司;ABI Step One Plus实时聚合酶链式反应(polymerase chain reaction,PCR)系统 美国Life Technologies公司;Illumina Mi Seq PE300高通量测序平台 美国Illumina公司;ND-2000C微量紫外分光光度计 美国Nano Drop公司。
1.3 细菌测序
对细菌序列16S rDNA V3~V4区域进行相关测序。细菌16S rDNA V3~V4区应用的引物为515F(5′-GTGYCAGCMGCCGCGGTAA-3′)、806R(5′-GGACTACHVGGGTWTCT-3′)。将稀释后的DNA基因组作为模板,使用带Barcode的特异引物、Phusion?High-Fidelity PCR Master Mix with GC Buffer和高保真酶进行PCR扩增,确保扩增的效率和准确性。扩增条件:94 ℃、2 min,然后98 ℃、10 s,62 ℃、30 s,68 ℃、30 s进行30 个循环,最后68 ℃、5 min。PCR产物用2%电泳凝胶进行检测。扩增体系(50 μL):5 μL 10×KOD缓冲液、5 μL 2 mmol/L dNTPs、3 μL 25 mmol/L MgSO4、上下游引物各1.5 μL(10 μmol/L)、1 μL DNA聚合酶、100 ng模板DNA。然后使用1×TAE 2%琼脂糖凝胶电泳纯化PCR产物,切出目标条带并对其进行回收。文库建成并测试合格后,上机测序。
1.4 数据分析及图像处理
1.4.1 细菌群落α多样性分析
α多样性是反映物种丰富度和同质性的综合指标,可用于分析样本的复杂性,即样本中群落的多样性。利用Spss软件对实验数据进行聚类分析。其中包括Observed-otus、Chao1指数、Shannon指数、Simpson指数、ACE指数和覆盖率。
1.4.2 细菌群落组成分析
采用RDP分类算法选择其中最常见的序列作为代表操作分类单元(operational taxonomic units,OTUs)的序列。OTUs序列的物种注释,用Mothur与SILVA132[21]的SSUrRNA数据库[22]进行设定阈值为0.8~1.0的物种注释剖析,在每个分类层次计算每个样本的界、门、纲、目、科、属、种水平群落组成。使用MUSCLE(版本3.8.31)软件[23]对多个序列进行快速测序,可以得到所有OTUs序列的系统发生关系。最后,每个样本的数据都是平均值,以数据量最少的样品为标准进行均一化处理,样本中的最小数据量为标准。随后的α多样性分析和β多样性分析基于均值数据。按照分析结果,绘制样品的稀释曲线、热图、菌群分布直方图和高质量序列分布图。
2 结果与分析
2.1 不同加工阶段腊肉中细菌测序结果分析
对徽派腊肉中提取的微生物总DNA进行V3~V4区间的高通量测序,在优化软件数据后,对排序结果进行排序和过滤。共获得1 202 200 个高质量序列,由表2可知,有效序列平均长度为253 bp。序列长度与所设计出的引物序列长度比较接近,说明腊肉样本的测序结果比较合理,可以满足后续的数据分析。
2.2 不同加工阶段腊肉中细菌多样性分析
2.2.1 不同加工阶段腊肉中细菌群落α多样性分析
α多样性主要指一定区域或特定生态系统本身的多样性,是一种能够反映物种丰富度和同质性的综合性质指标。α多样性一方面与菌群丰富度有关,另一方面与菌群多样性有关。菌群丰富度指数主要包括ACE指数和Chao1指数。菌群多样性指数主要包括覆盖率和Shannon指数。Chao1指数可以估计微生物群落中的OTU数量,并计算微生物群落的丰度。通常而言,Chao1指数越大,样品中的微生物群落物种总数越多。
由表3可知,在加工前期(鮮肉、腌制中期、腌制结束、成熟中期),Chao1指数较大,而成熟结束,Chao1指数明显减小。ACE指数表现出与Chao1指数类似的变化规律,说明加工后期腊肉样品中细菌群落丰富度变化较大。Shannon指数和Simpson指数可以表征微生物的丰度和同质性。Shannon指数越高,微生物群落的多样性就越大。Shannon指数和Simpson指数呈先升高后降低的趋势,说明腊肉样品中细菌群落多样性随发酵时间的延长发生了显著变化,在加工后期,微生物多样性降低。覆盖率可以反映测序深度,覆盖率越接近1,测序越合理。本研究中所有样品覆盖率均大于0.997,说明徽派腊肉样品中的序列基本检测到,测序结果可以反映徽派腊肉样品中微生物的真实状况。
2.2.2 不同加工阶段腊肉中细菌稀释曲线
稀释和等级聚类曲线是用来描述样品中微生物群落多样性的曲线。稀释曲线可以直接反映测序量的合理性,也可以间接反映样品中物种的数量。由图1可知,所有徽派腊肉样品的稀释曲线趋向平坦,表明测序深度合理。随着测序深度的增加,加工过程中的各个阶段腊肉样本的稀释曲线呈现先上升后趋于平缓的趋势。表示样品选定的序列数据足够多,可以反映样品中大多数微生物区域的数据,测序结果合理。
等级聚类曲线是样本中按相对丰度(或可用序列数)从大到小排序得到的对应排序编号。在等级聚类曲线中,横坐标方向上,曲线的宽度反映了物种的丰富性。纵坐标方向上,样品中物种均匀度由曲线的平滑程度来表示。如果曲线显示平滑,说明物种是均匀分布的[24]。由图2可知,随着腊肉加工各个阶段的加工时间增加,曲线呈现先下降后趋于平缓的趋势,说明在测序过程中物种分布较为均匀。
2.2.3 不同加工阶段腊肉中细菌群落β多样性分析
由图3可知,徽派腊肉整个加工过程中,微生物的种间差异逐渐增加,在成熟后期,各物种间的差异达到最大。
2.3 不同加工阶段腊肉中物种分布情况
2.3.1 物種相对丰度
在门分类水平上,共有47 个细菌门被检出,选取丰度排名前10的细菌绘制相对丰度柱形图。由图4可知,厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、放线菌门(Actinomycetes)、拟杆菌门(Bacteroides)占主导地位。在整个加工过程中,厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)具有主要优势。其中厚壁菌门(Firmicutes)相对丰度最大,鲜肉、腌制中期、腌制结束、成熟中期和成熟结束的相对丰度分别为51.1%、38.2%、39.4%、38.8%和52.5%。其次,变形菌门(Proteobacteria)是另一优势微生物,鲜肉、腌制中期、腌制结束、成熟中期和成熟结束的相对丰度分别为33.7%、45.3%、39.0%、52.2%和42.8%。此外,放线菌门(Actinomycetes)在鲜肉、腌制中期、腌制结束、成熟中期和成熟结束的相对丰度分别为3.5%、3.9%、4.3%、1.3%和1.6%,而拟杆菌门(Bacteroides)在鲜肉、腌制中期、腌制结束、成熟中期和成熟结束的相对丰度分别为7.8%、9.3%、14.2%、5.7%和2.3%。
在属分类水平上,样品中共检测到676 种细菌,选取丰度排名前30的细菌绘制相对丰度柱形图。由图5可知,在整个加工过程中,葡萄球菌(Staphylococcus)、乳球菌(Lactococcus)、链球菌(Streptococcus)、嗜冷杆菌(Psychrobacter)、盐弧菌(Salinivibrio)和不动杆菌(Acinetobacter)的相对丰度较大,是优势细菌。李彦虎[25]研究发现,陇西腊肉中主要细菌是环丝菌(Cyclomycetes)、肉杆菌(Botulinum)、乳球菌、假单胞菌(Pseudomonas)和嗜冷杆菌。董蕴等[26]研究发现,湖北恩施腊肉的主要细菌种类为葡萄球菌、嗜冷杆菌、假单胞菌、环孢菌(Cyclospora)、盐单胞菌(Cobetia)和不动杆菌。Cordero等[27]发现,使用食盐的西班牙火腿中60%微生物为葡萄球菌。Rodríguez[28]、Lorenzo[29]等分别对意大利和西班牙火腿进行研究,在发酵过程中与发酵后期,葡萄球菌和微球菌(Micrococcus)数量明显上升,与本研究结果相似。金黄色葡萄球菌(Staphylococcus aureus)有抑制腐败细菌生长的作用,并在发酵中起良好作用[30]。由于徽派腊肉在发酵阶段均处于低温状态,导致大量嗜低温菌与耐盐菌大量繁殖。而在腊肉的发酵过程中,样品中的细菌明显出现演替现象。例如,葡萄球菌相对丰度由鲜肉的0.3%到成熟结束时增加至44.2%,达到最高,可能是由于葡萄球菌对于环境因子的耐受性较高,葡萄球菌同时也是腊肉风味的来源之一。不动杆菌相对丰度呈现先增加后降低的趋势,在成熟中期时达到最高(16.6%),在成熟结束时降低至7.8%。盐单胞菌属(Halomonassp)(属于图中Others分类)在腌制过程中也呈现一直增加的趋势,在成熟结束时达到最高(6.9%)。
2.3.2 物种丰度聚类热图
为更直观表现徽派腊肉发酵过程中优势细菌的丰度变化,挑选相对丰度排名前35的属绘制热图。
由图6可知,鲜肉优势菌为乳酸菌(Lactobacillus)、棒状杆菌(Corynebacterium)、肉杆菌(Carnobacterium)和肠球菌(Enterococcus)等。腌制中期的主要细菌为肠杆菌(Enterobacteriaceae)。腌制结束的优势菌主要为双歧杆菌(Bifidobacterium)、链球菌(Streptococcus)和嗜冷杆菌。成熟中期样品的优势菌主要为嗜冷细菌(Psychrophilic bacteria)、弧菌(Vibrio),成熟结束,弧菌(Vibrio)、耐盐菌和葡萄球菌(Staphylococcus)最为显著。
在腌制阶段,由于环境温度较低,渗透压较高,使得这个阶段的优势菌以耐盐菌与嗜冷菌为主。到了成熟阶段,主要的变化以水分含量为主,并且不同类型的微生物相互抑制,使腊肉样品中的菌群得到进一步优化。陈竞适等[31]在湘西腊肉中发现了微球菌、乳杆菌及葡萄球菌。Wang Xinghong[32]、Wang Xinhui[33]等在宣威火腿中发现了微球菌、棒状杆菌和乳酸杆菌,在四川腊肉中发现了葡萄球菌、乳球菌和芽孢杆菌。Papamanoli等[34]对广式腊肠进行研究,发现主要微生物为乳杆菌、片球菌和葡萄球菌,与本研究较为一致。
2.3.3 LEfSe分析
LEfSe分析(线性判别分析(Linear Discriminant Analysis,LDA)对数评分阈值≥4)展现的是有显著性差异的物种,本研究用于分析明确不同阶段处理过程中群落组成的异同。由图7可知,鲜肉、成熟中期和成熟结束时,样品中共有24 个明显不同的类群。其中,成熟结束时的细菌群落明显比鲜肉和腌制结束时多,并以弧菌、葡萄球菌和耐鹽菌为主。
由图8可知,在鲜肉、成熟中期和成熟结束时,分别观察到共有9、3、12 个分类群存在显著差异,表明随着生产过程的进行,不同加工时间的群落组成相似度先减小后增加,说明在加工后期腊肉中逐渐形成优势菌。
3 结论
本研究采用HTS技术,对徽派腊肉中的微生物群落构成进行研究。结果表明:在腊肉发酵过程中,微生物群落发生较大变化,优势菌由乳球菌(Lactococcus)、双歧杆菌(Bifidobacterium)和芽孢杆菌(Bacillus)等逐渐演替为嗜冷杆菌(Psychrobacter)、弧菌(Vibrio)、盐单胞菌(Halomonas)和葡萄球菌(Staphylococcus);发酵时间越久,菌群分布越稳定,优势菌越明显。因此,对徽派腊肉进行微生物测序研究有助于解析其独特风味的来源,同时为徽派腊肉生产标准的制定等提供理论依据。
参考文献:
[1] 柴子惠. 低钠腊肉加工和贮藏期间理化特性和菌相变化的研究[D]. 重庆: 西南大学, 2019.
[2] GUO Xin, HUANG Feng, ZHANG Hong, et al. Classification of traditional Chinese pork bacon based on physicochemical properties and chemometric techniques[J]. Meat Science, 2016, 117: 182-186. DOI:10.1016/j.meatsci.2016.02.008.
[3] 贺雪华, 李林, 白登荣, 等. 改进型城口腊肉贮藏过程中的品质变化及货架期预测[J]. 食品科学, 2017, 38(11): 249-255. DOI:10.7506/spkx1002-6630-201711040.
[4] 粟桂蓉, 彭钰媛, 周璐璐, 等. 传统土家腊肉加工过程中风味物质研究[J]. 食品科技, 2017, 42(3): 118-123.
[5] KO?O?YN-KRAJEWSKA D, DOLATOWSKI Z J. Probiotic meat products and human nutrition[J]. Process Biochemistry, 2012, 47(12): 1761-1772. DOI:10.1016/j.procbio.2012.09.017.
[6] COMI G, ANDYANTO D, MANZANO M, et al. Lactococcus lactis and Lactobacillus sakei as bio-protective culture to eliminate Leuconostoc mesenteroides spoilage and improve the shelf life and sensorial characteristics of commercial cooked bacon[J]. Food Microbiology, 2016, 58: 16-22. DOI:10.1016/j.fm.2016.03.001.
[7] ZHAO C J, SCHIEBER A, GAENZLE M G. Formation of taste-active amino acids, amino acid derivatives and peptides in food fermentations: a review[J]. Food Research International, 2016, 89: 39-47. DOI:10.1016/j.foodres.2016.08.042.
[8] YU Hai, YIN Yongqi, XU Lin, et al. Genome sequence of Klebsiella pneumoniae YZUSK-4, a bacterium proposed as a starter culture for fermented meat products[J]. Genome Announcements, 2015, 3(4): e00774-15. DOI:10.1128/genomea.00774-15.
[9] ZHAO Changqing, LU Ziyang, HUANG Jing, et al. Optimum for pork jerky fermented with Lactobacillus acidophilus[J]. Journal of Biobased Materials and Bioenergy, 2017, 11(1): 21-26. DOI:10.1166/jbmb.2017.1640.
[10] BERDAGU? J L, MONTEIL P, MONTEL M C, et al. Effect of starter cultures on the formation of flavor compounds in dry sausage[J]. Meat Science, 1993, 35(3): 275-287. DOI:10.1016/0309-1740(93)90033-E.
[11] BRUNA J M, HIERRO E, DE LA HOZ L, et al. Changes in selected biochemical and sensory parameters as affected by the superficial inoculation of Penicillium camemberti on dry fermented sausages[J]. International Journal of Food Microbiology, 2003, 85(1/2): 111-125. DOI:10.1016/S0168-1605(02)00505-6.
[12] CHEN K, PACHTER L. Bioinformatics for whole-genome shotgun sequencing of microbial communities[J]. PLoS Computational Biology, 2005, 1(2): 106-112. DOI:10.1371/journal.pcbi.0010024.
[13] TRINGE S G, RUBIN E M. Metagenomics: DNA sequencing of environmental samples[J]. Nature Reviews Genetics, 2005, 6(11): 805. DOI:10.1038/nrg1709.
[14] ZHU Qianglong, LIU Shi, GAO Peng, et al. High-throughput sequencing technology and its application[J]. Journal of Northeast Agricultural University (English Edition), 2014, 21(3): 84-96. DOI:10.1016/S1006-8104(14)60073-8.
[15] YANG Lin, YANG Huilin, TU Zongcai, et al. High-throughput sequencing of microbial community diversity and dynamics during Douchi fermentation[J]. PLoS One, 2016, 11(12): e0168166. DOI:10.1371/journal.pone.0168166.
[16] LIANG Huipeng, YIN Liguo, ZHANG Yahao, et al. Dynamics and diversity of a microbial community during the fermentation of industrialized Qingcai paocai, a traditional Chinese fermented vegetable food, as assessed by Illumina MiSeq sequencing, DGGE and qPCR assay[J]. Annals of Microbiology, 2018, 68(2): 111-122. DOI:10.1007/s13213-017-1321-z.
[17] YANG Hongyan, WU Hao, GAO Lijuan, et al. Effects of Lactobacillus curvatus and Leuconostoc mesenteroides on Suan cai fermentation in Northeast China[J]. Journal of Microbiology and Biotechnology, 2016, 26(12): 2148-2158. DOI:10.4014/jmb.1607.07010.
[18] WANG Xinghui, WANG Songhu, ZHAO Hai. Unraveling microbial community diversity and succession of Chinese Sichuan sausages during spontaneous fermentation by high-throughput sequencing[J]. Journal of Food Science and Technology-Mysore, 2019, 56: 3254-3263. DOI:10.1007/s13197-019-03781-y.
[19] 文开勇, 汪月, 文鹏程, 等. 四川传统腊肉中微生物群落结构研究[J]. 食品与发酵工业, 2020, 46(3): 36-42. DOI:10.13995/j.cnki.11-1802/ts.022133.
[20] 毕旺来, 胡雷凤, 明亮, 等. 烟熏腊肉货架期主要微生物的研究[J]. 武汉轻工大学学报, 2019, 38(2): 16-20. DOI:10.3969/j.issn.2095-7386.2019.02.003.
[21] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10(10): 996. DOI:10.1038/NMETH.2604.
[22] WANG Q G, GARRITY M, TIEDJE J M, et al. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267. DOI:10.1128/AEM.00062-07.
[23] QUAST C, PRUESSE E, YILMAZ P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools[J]. Nucleic Acids Research, 2013, 41: D590-6. DOI:10.1093/nar/gks1219.
[24] LUNDBERG D S, YOURSTONE S, MIECZKOWSKI P, et al. Practical innovations for high-throughput amplicon sequencing[J]. Nature Methods, 2013, 10(10): 999. DOI:10.1038/nmeth.2634.
[25] 李彦虎. 传统陇西腊肉制作过程中微生物群落演替与腊肉风味的相关性分析[D]. 兰州: 甘肃农业大学, 2020.
[26] 董蕴, 王玉荣, 王尧, 等. 基于变性梯度凝胶电泳和MiSeq高通量测序技术分析恩施地区腊肉的细菌多样性[J]. 肉类研究, 2018, 32(10): 37-42. DOI:10.7506/rlyj1001-8123-201810007.
[27] CORDERO M R, ZUMALAC?RREGUI J M. Characterization of micrococcaceae isolated from salt used for Spanish dry-cured ham[J]. Letters in Applied Microbiology, 2000, 31(4): 303-306. DOI:10.1046/j.1472-765x.2000.00818.x.
[28] RODR?GUEZ M, N??EZ F, C?RDOBA J J, et al. Characterization of Staphylococcus spp. and Micrococcus spp. isolated from Iberian ham throughout the ripening process[J]. International Journal of Food Microbiology, 1994, 24(1/2): 329-335. DOI:10.1016/0168-1605(94)90131-7.
[29] LORENZO J M, GARC?A FONT?N M C, G?MEZ M, et al. Study of the Micrococcaceae and Staphylococcaceae throughout the manufacture of dry-cured Lacón (a Spanish traditional meat product) made without or with additives[J]. Journal of Food Research, 2012, 1(1): 200-211. DOI:10.5539/jfr.v1n1p200.
[30] 李珊珊, 祝超智, 崔文明, 等. 發酵肉制品中微生物发酵剂分离筛选及应用研究进展[J]. 肉类研究, 2019, 33(7): 61-66. DOI:10.7506/rlyj1001-8123-20190424-089.
[31] 陈竞适, 刘静, 任海姣, 等. 湘西陈年腊肉微生物群落分析及高产脂肪酶细菌的筛选[J]. 肉类研究, 2017, 31(3): 1-6. DOI:10.7506/rlyj1001-8123-201703001.
[32] WANG Xinghong, MA Ping, JIANG Dongfu, et al. The natural microflora of Xuanwei ham and the no-mouldy ham production[J]. Journal of Food Engineering, 2006, 77(1): 103-111. DOI:10.1016/j.jfoodeng.2005.06.047.
[33] WANG Xinhui, ZHANG Yalin, REN Hongyang. Effects of grape seed extract on lipid oxidation, biogenic amine formation and microbiological quality in Chinese traditional smoke‐cured bacon during storage[J]. Journal of Food Safety, 2018, 38(2): e12426. DOI:10.1111/jfs.12426.
[34] PAPAMANOLI E, KOTZEKIDOU P, TZANETAKIS N, et al. Characterization of Micrococcaceae isolated from dry fermented sausage[J]. Food Microbiology, 2002, 19(5): 441-449. DOI:10.1006/fmic.2002.0503.
收稿日期:2020-12-15
基金項目:中央高校基本科研业务费专项资金项目(JZ2020HGQA0150);
