Spatial variations of bacterial community composition in sediments of the Jiaozhou Bay, China*
2021-06-15QiqiSUNJinmingSONGXuegangLIHuamaoYUANJianweiXING
Qiqi SUN, Jinming SONG ,4,**, Xuegang LI ,4, Huamao YUAN ,4, Jianwei XING
1 Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences,Qingdao 266071, China
2 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology(Qingdao), Qingdao 266071, China
3 Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao 266071, China
4 University of Chinese Academy of Sciences, Beijing 100049, China
Abstract Spatial variations of sediment microbes pose a great challenge for the estimation of anthropogenic influence on biogeochemical processes, yet remain very unclear in coastal ecosystems.Surface sediments in 9 stations from the eutrophic Jiaozhou Bay, China, were sampled, DNA was extracted within the sediments, and the 16S rDNA was sequenced with the Illumina Hiseq sequencing. Results reveal considerable heterogeneity of sediment bacteria in the Jiaozhou Bay, of which Proteobacteria and Bacteroidetes accounted for over 75%. Bacterial alpha-diversity indices decreased generally from the outside to the inner part of the bay and from the off shore to the nearshore area. Bacterial community structures of S3, S4, S7, and S8 clustered, those of S5, S13, and S14 grouped together, while those of S6 and S10 were distinct from each other and from those of the other stations. Major class Gammaproteobacteria were more abundant at the stations with mesoeutrophic to eutrophic levels (S4, S5, S8, and S10) and less abundant at oligotrophic stations (S6, S13, and S14), while Deltaproteobacteria had an opposite distribution pattern.Overall, bacterial community composition transitioned from being Xanthomonadales-dominant at S4 and S8 to being unidentifed_Gammaproteobacteria-dominant at S5, S6, S13, and S14, while in other stations there were comparable orders. The biogeochemical processes correspondingly changed from being nitrogen cycling-dominant at S4 and S8 to being sulfur cycling-dominant at S5, S6, S13, and S14. The bacterial distribution patterns were especially aff ected by the factors (dissolved organic phosphorus, DOP) in the overlying seawater due to the habitat status of P-insuffi ciency in the bay. Both orders Xanthomonadales and Alteromonadales could serve as bioindicators of anthropogenic pollution to diff erent pollution types. At last,divergent distribution patterns ofindividual bacterial populations in the bay were revealed, the influential environmental gradients were clarified, and the uncertainty of microbes was reduced, helping to predict environmental functions in coastal areas.
Key word: Jiaozhou Bay; bacterial community; spatial variation; sediments; overlying water
1 INTRODUCTION
Microbes are the engine that drives global biogeochemical cycling (Azam and Malfatti, 2007).Sediment microbes (especially bacteria) are a key part of marine ecosystems, which play an important role in regulating the transformations of various biogenic elements (Yokokawa and Nagata, 2010; Sun et al., 2011). For a certain ecosystem, sediment bacterial communities are influenced by their overlying seawater habitats (Stieglmeier et al., 2014).As a complex ecosystem connecting terrestrial and marine habitats (Su et al., 2005), coastal regions(<200 m in depth) are susceptible to more intensive terrestrial inputs and artificial disturbances (Nogales et al., 2011). These ecosystems always harbor microbial communities in certain degree of spatial heterogeneity due to their large gradients in nutrient concentrations and relevant parameters, etc. (Hewson and Fuhrman, 2004). Such heterogeneity of bacterial communities can lead to additional uncertainty in accurately predicting ecosystem responses to environmental changes and providing better ecosystem services (Kieft et al., 2018). In addition,bacterial communities have served as sensitive indicators of eutrophication and contamination in coastal aquatic sediments due to their sensitivity to environmental changes (Aylagas et al., 2017; Simonin et al., 2019). Considering their key role in ecological function and environmental sustainability, further research is needed regarding bacterial communities in coastal ecosystems and major variables shaping their distribution patterns.
At present, spatial variations of microbes in coastal ecosystems and how they indicate the structure and functioning of marine ecosystems have gained growing attention (Fuhrman et al., 2015), yet the research results varied with ecosystems. It has been widely known that bacterial community composition is more complex in coastal areas such as the Bohai Sea than in open seas such as the northern Yellow Sea(Yu et al., 2018). In recent years, obvious spatial variations have been observed successively in the Jiaozhou Bay (JZB) regarding the sediment ammoniaoxidizing Crenarchaeota (Dang et al., 2008),NirSencoding bacterial assemblages (Dang et al., 2009),ammonia-oxidizing Betaproteobacteria (Dang et al.,2010a) and anammox bacterial communities (Dang et al., 2010b), etc. Research on the sediment bacterial community of four stations in the JZB showed that it was the geographic heterogeneity rather than seasons that determined the benthic microbiomes (Liu et al.,2015). The bacterial community in the Yellow Sea exhibited relatively low spatial variations among the four diff erent coastal sites characterized by rocky,sandy, muddy, and artificial harbors, respectively(Wang et al., 2009). In addition, the negligible spatial variation of bacterioplankton community has been also observed in the Chesapeake Bay (Kan et al.,2007). Usually, the heterogeneity ofindividual bacterial populations is accompanied by their functional roles and their relative influence on biogeochemical cycling (Kieft et al., 2018).Understanding the bacterial community composition and their influencing factors is key to the accurate prediction of the ecosystem responses to environmental changes and thus providing better ecosystem services.
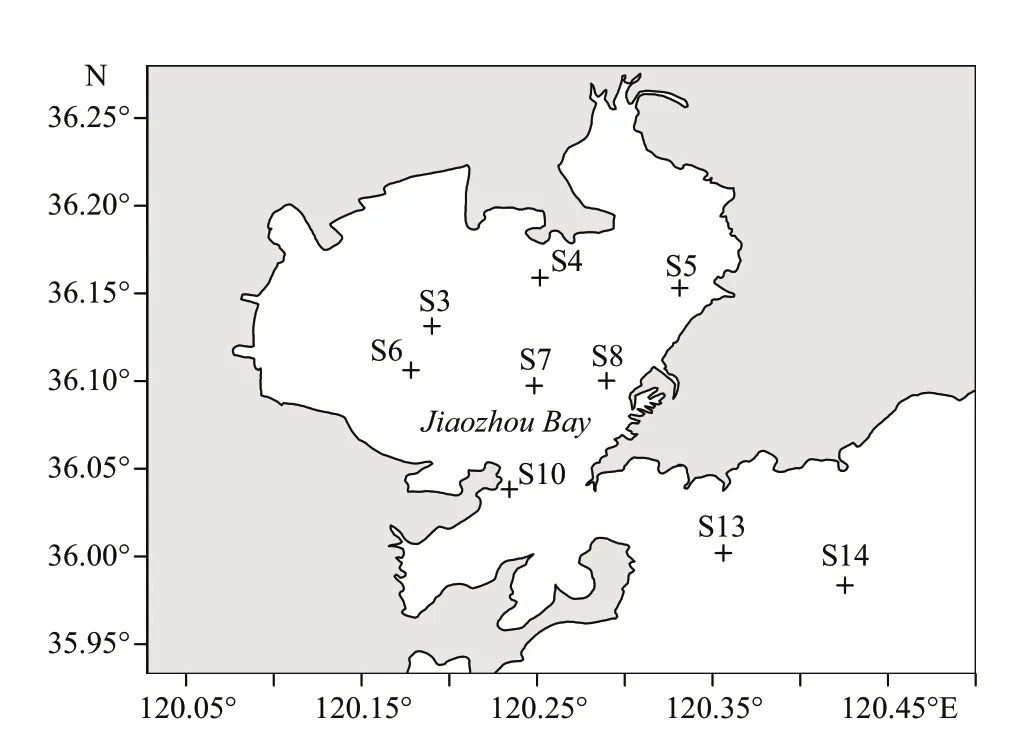
Fig.1 Location of the JZB and sampling stations in November 2019
In this study, bacterial distribution at 9 stations in the JZB was investigated within the surface sediments,with nutrients and relevant parameters determined in the sediments and the overlying seawater. The aims were to present the distribution patterns in bacterial communities of the bay; clarify the influential environmental gradients on the spatial variations of sediment bacterial community composition; and explore possible indicating functions of specific taxa among the bacterial communities.
2 MATERIAL AND METHOD
2.1 Site description and sediment sampling
The JZB (Fig.1) is a large semi-enclosed water body located on the western coast of the Yellow Sea and exchanges with the open sea via a 2.5-km wide mouth (Dai et al., 2007). The mouth of the JZB is narrow and the water-exchange between the inside and the outside of the bay is poor which may constrain the diff usion of the pollutants and the self-purification of the bay. The shallow water column (7 m in average)and JZB tides jointly result in vertical profiles of temperature, turbidity, pH, and dissolved oxygen that were nearly homogeneous. However, the salinity was the greatest at S14 (Table 1). No less than 10 small rivers enter the bay carrying diff erent amounts of sediment and pollutants, especially the Yangge, Dagu,Moshui, Loushan, Licun, and Haibo Rivers (Liu et al., 2011). In recent 30 years, the JZB has experienced various anthropogenic impacts and diff erent eutrophication gradients (Yuan et al., 2016), providingan ideal model system for the study of the distribution pattern of coastal microorganisms and related ecological variables.
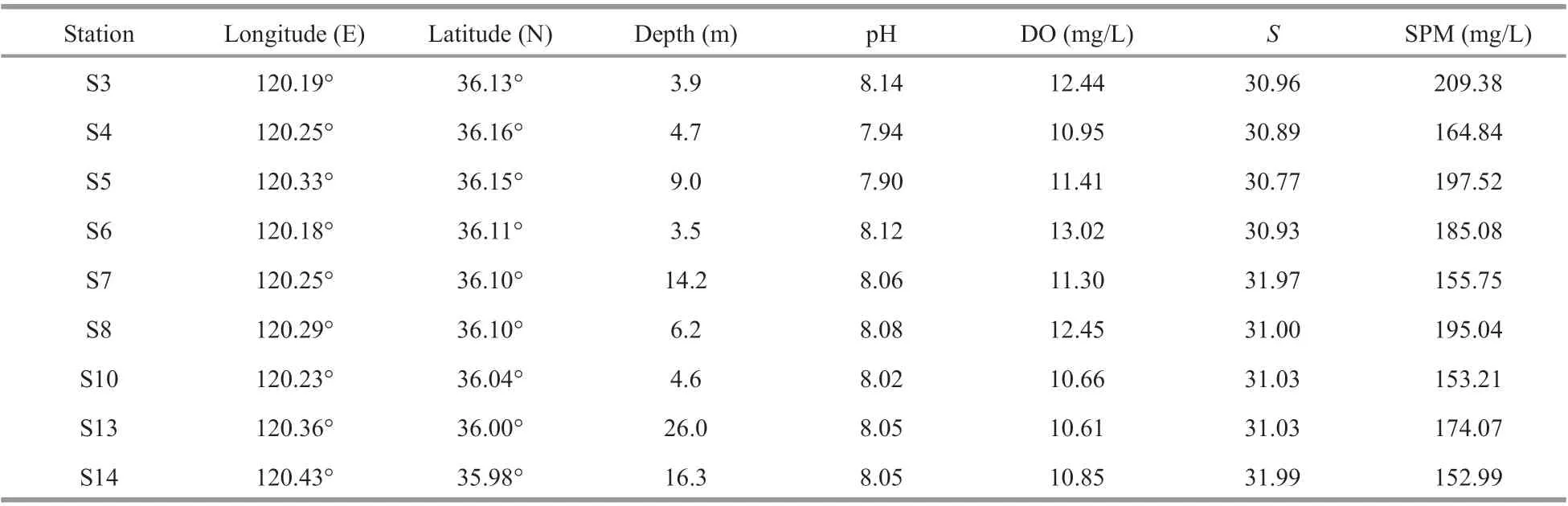
Table 1 Basic information ofindividual stations
The field observation was carried out in the JZB cruise onboard the R/VChuangxin(innovation in Chinese) on November 18–19, 2018. During the cruise, 9 stations were deployed over the entire bay and sampled (Fig.1): stations S3, S4, S5, S6, S7, and S8 are located inside the bay, S10 is at the bay mouth,and S13 and S14 are located outside the bay. Basic information of the sampling stations is shown in Table 1. The 9 stations were grouped according to the pollution degree: S4, S5, and S10 are heavily polluted,S3, S6, S7, and S8 are less polluted, and S13 and S14 are aff ected by seaport and mildly polluted (Liu et al.,2015). It is noteworthy that S10 is the most seriously oil-contaminated station due to the frequent oil-spill events (Dang et al., 2010a) and an anticlockwise eddy, resulting in water quality worsening (Dang et al., 2009). Surficial sediments were sampled using a Van Veen grab sampler and then transported to the laboratory. All sediment samples were deep-frozen immediately after being transferred into the laboratory in 72 h, lyophilized and then ground for chemical analysis.
Seawater at 2 m above the bottom was sampled with 10-L Niskin bottles at each station and analyzed for nutrients and chlorophylla(Chla) (Yuan et al.,2018). After collection, seawater samples were filtered through 47-mm Whatman GF/F filters that were pre-combusted at 450 °C for 4 h and washed(1-mol/L HCl). The filtrates were stored in acidcleaned HDPE bottles at -20 °C and then brought to the laboratory for dissolved nutrient analysis. The filters were rinsed with distilled water to remove dissolved nutrients after filtration, and then frozen at-20 °C for particulate nutrients analysis. About 200-mL seawater was filtered through 0.45-μm pore-size acetate cellulose filter under low vacuum and then preserved at -20 °C for the Chlaanalysis. The pH,salinity (S), and dissolved oxygen (DO) of the overlying water were measured using a Multi-function Measuring Instrument (Orion, Versa Star, Thermo Fisher Inc., USA) (Budhavant et al., 2014).
2.2 Nutrient determination
Inorganic nitrogen (IN) was extracted in 25-mL 0.1-mol/L HCl for 2 h, with the residue from IN extraction being treated with alkaline K2S2O8at 124 °C for 1 h to digest organic nitrogen (ON) (Liu et al.,2002). Ammonia (NH4-N), nitrate (NO3-N) and nitrite(NO2-N) in the HCl and K2S2O8extracts were then determined photo-metrically following the standard procedure using a continuous flow analyzer (QuAAtro,Bran-Luebbe Inc., Germany) (Xing et al., 2017), the sum of them was IN. Total phosphorus (TP) and inorganic phosphorus (IP) were determined based on HCl-extractable P (1-mol/L HCl, 24 h) of combusted(550 °C, 2 h) and non-combusted sediments, respectively(Aspila et al., 1976), while organic phosphorus (OP)was the diff erence of TP and IP. A strong alkaline leaching method was adopted for the determination of the biogenic silica (BSi) (Mortlock and Froelich,1989) and the concentrations of BSi were then converted to weight per mille SiO2of dry bulk sediments with the slope for mineral correction(DeMaster, 1981, 1991). The detection limits for NH4-N, NO3-N, NO2-N, dissolved inorganic phosphorus (PO4-P) and dissolved inorganic silicate(SiO3-Si) were 0.25, 0.40, 0.01, 0.03, and 0.01 μmol/L,respectively. National standard references were used to satisfy the precision and accuracy. The precisions and recoveries for all nutrient species were better than 5% and between 95.0% and 105.1%, respectively.Chlaon the cellulose-acetate membranes was extracted with 10-mL N, N-dimethylformamide at 4 °C in darkness for 0.5 h and then determined using a fluorescence spectrophotometer (Hitachi F-4600),with the variation coeffi cient<5% (Suzuki and Ishimaru, 1990).
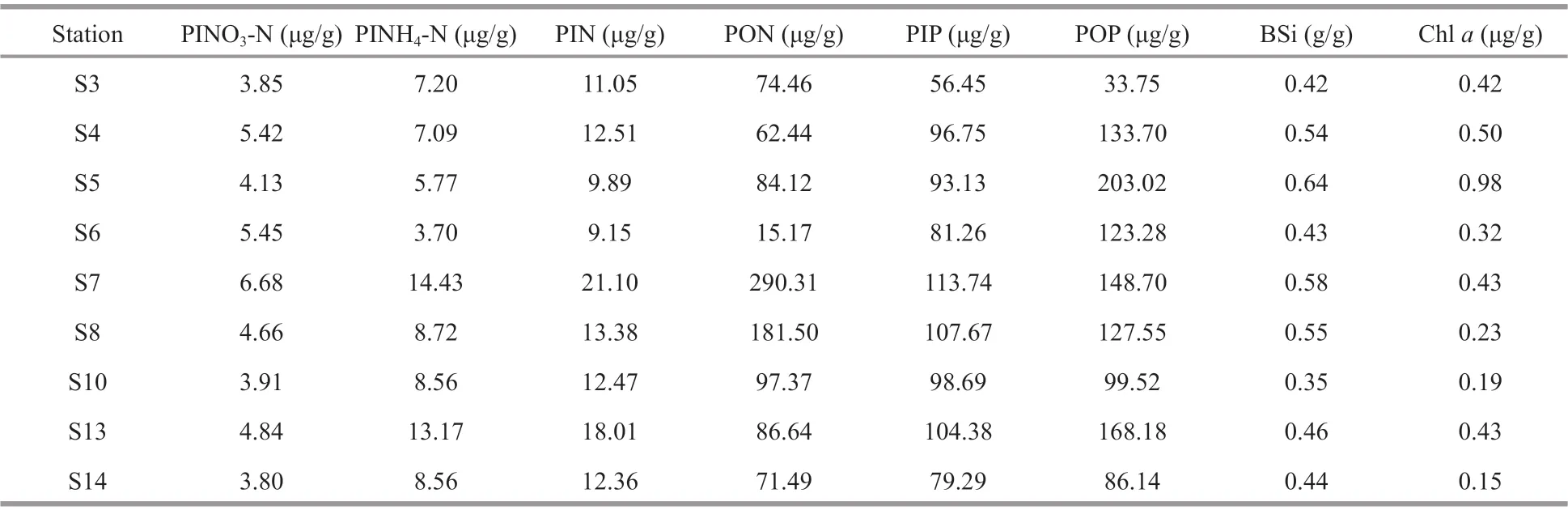
Table 2 Nutrient contents in the surface sediments of the JZB
2.3 DNA extraction and Illumina HiSeq highthroughput sequencing
Total genomic DNA of all sediment examples was extracted using the CTAB/SDS method with Qiagen Gel Extraction Kit (Qiagen, Germany) following the manufacturer’s recommendations, with the hypervariable V4 region of the 16S rRNA gene amplified from the bacteria (Caporaso et al., 2011).Sequencing libraries were generated using TruSeq®DNA PCR-Free Sample Preparation Kit (Illumina,USA) and library quality was tested in the Qubit®2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. Sequencing was conducted on an Illumina HiSeq 2500 platform (Illumina Corporation, San Diego, USA). Approximately 80 117 high-quality prokaryotic sequences per sample with the average length of approximately 420 bp were produced.
2.4 Data analysis
UPARSE software package with the UPARSEOTU and UPARSE-OTUref algorithms was used in sequence analysis and operational taxonomic units(OTUs) clustering (Edgar, 2013). Taxonomy was assigned using the Ribosomal Database Project classifier (Wang et al., 2007). Each sample was rarefied to the same number of reads (80 117 sequences) for both alpha-diversity and beta-diversity analysis.
Principal Coordinate Analysis (PCoA) was conducted to test the similarity of bacterial community structure of the samples. Pearson correlation analysis was carried out to test the correlations between bacterial communities and influential parameters.Redundancy analysis (RDA) was performed to further identify the influential variable gradients of bacterial groups. Pearson correlation analysis was performed using SPSS 20.0 software (SPSS Inc., Chicago,USA), withP<0.05 (two-tailed) being set as the significant level. Analyses of PCoA and RDA were performed with the R software package V.3.6.1(vegan, ape, ggplot). Surfer 15.0 was used to draw the sampling location map and isolines of bacterial classes in the JZB, with the “Kriging” interpolation method selected. The cumulative histogram was generated using Sigmaplot 12.5 software (Systat Software Inc., San Jose, CA, USA).
3 RESULT
3.1 Nutrients in the sediments and in the overlying water
For most stations, particle organic nitrogen (PON)and particle organic phosphorus (POP) were the main components of total nitrogen (TN) and total phosphorus (TP) in the sediments, while particle inorganic nitrogen (PIN) was characterized by particle ammonia nitrogen (PINH4-N) (Table 2). The nutrient contents in the sampled sediments of the JZB varied with stations. Nutrients such as the particle nitrate nitrogen (PINO3-N), PINH4-N, PIN, PON, and particle inorganic phosphorus (PIP) in the sediments were the greatest at S7 (at the center of the inner bay),while POP, BSi, and Chlahad the maximum value at S5 (near the Licun River). The least values ofPINH4-N, PIN, and PON was recorded at S6 (in the west part of the inner bay), while PIP and POP at S3(near S6) had the least values. BSi was the least at S10 (at the mouth) and Chlawas the least at S14(outside the bay) (Table 2). BSi showed a positive correlation with Chla(Supplementary Table S1),indicating their common phytoplankton origins.
I made up my mind that I would save my paper delivery money, and in three weeks I had the seven dollars that I calculated would cover the cost of her window. I put the money in an envelope with a note explaining that I was sorry for breaking her window and hoped that the seven dollars would cover the cost for repairing it.
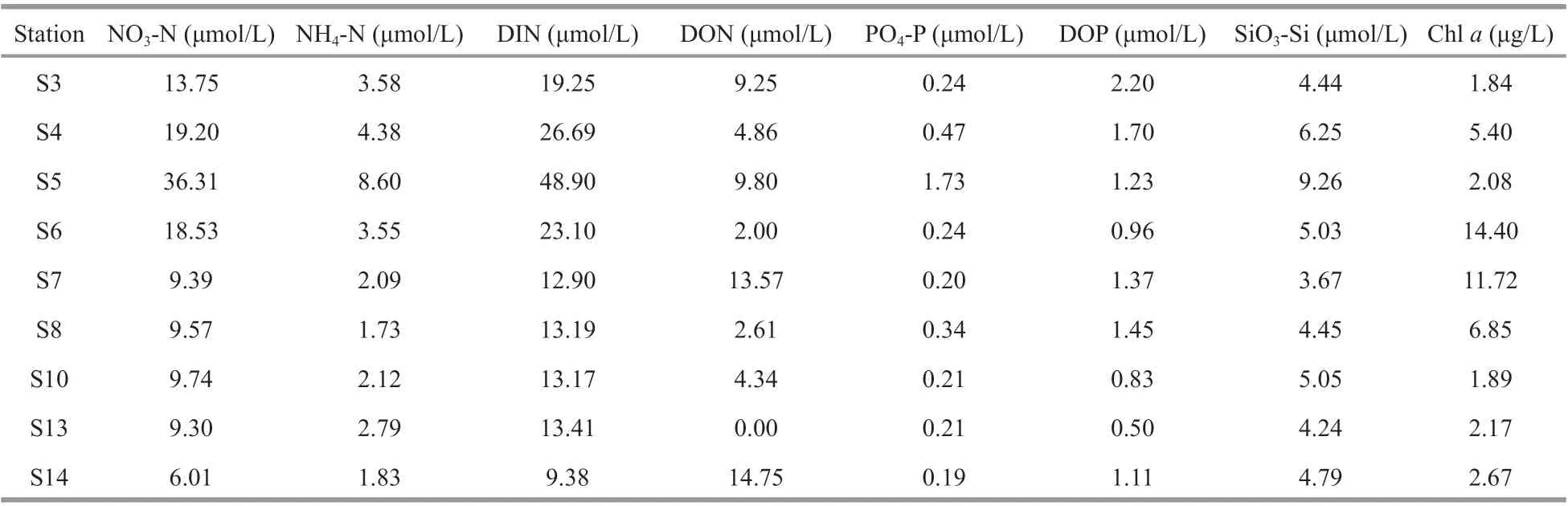
Table 3 Nutrient concentrations in the overlying seawater of the JZB
NO3-N (in average of 14.64 μmol/L) was the major form of dissolved inorganic nitrogen (DIN), followed by NH4-N (average of 3.41 μmol/L) (Table 3), which is in line with the findings of a previous study showing that the DIN composition of the JZB has changed from being NH4-N predominant to NO3-N predominant due to human intervention in recent years (Li et al., 2018). The overlying seawater of the sampling stations in the JZB also varied in trophic levels. S5 harbored the greatest NO3-N, NH4-N, DIN,PO4-P, and SiO3-Si, while S14 and S3 had the greatest dissolved organic nitrogen (DON) and dissolved organic phosphorus (DOP), respectively. S13 (outside the bay and near the mouth) had the least DON(0.00 μmol/L) and DOP (0.50 μmol/L), while S14(outside the bay and away from the mouth) had the least DIN (9.38 μmol/L) and PO4-P (0.19 μmol/L). S7 had the least SiO3-Si and the greatest Chlain the overlying water (Table 3), which corresponded well with the greatestS(Table 1). Correlation results showed that DIN, PO4-P, and SiO3-Si were correlated positively with each other (Supplementary Table S2),which indicated common origins and similar cycling processes (Yuan et al., 2018). Chlain the overlying water showed no correlation with dissolved nutrients.However, Chlain the sediment was significantly and positively correlated with DIN, PO4-P, and SiO3-Si(data not shown), which plausibly suggested the relevance of phytoplankton photosynthesis to the inorganic nutrient contents. The DIN/dissolved inorganic phosphorus (DIP) (ranging from 28.2 to 96.3) was greater than the Redfield ratio (16:1),indicating that the dissolved inorganic matter was more depleted in P than in N (Dortch and Whitledge,1992; Justić et al., 1995), which was consistent with the general DIP deficiency phenomenon compared with N in the JZB (Yuan et al., 2018).
3.2 Bacterial communities in sediment samples
There were in total 721 055 high-quality sequences for bacterial 16S genes of all sediment samples with an average of 80 117 sequences per sample.Eventually, the bacterial community had 59 929 clustered OTUs. In this study, the rarefaction curves of all the samples tended to be gentle (Fig.2a) with the coverage reaching up to over 99% (Table 4), which indicated that most species had been detected and the dataset could be used in the subsequent analysis. Both Chao1 richness and Shannon diversity indices varied with stations. Most stations had comparable Chao1 richness except stations S3, S7, S13, and S14 which had high values, and most stations had comparable Shannon diversity except stations S4, S8, and S7 which had low values. At most stations, Chao1 richness and Shannon diversity indices had consistent distribution patterns in the JZB, such as at stations S4 and S8 both values of which were lower and at stations S3, S13, and S14 both values of which were higher.Exceptionally, at S7 Chao1 richness was relative higher but Shannon diversity was lower (Table 4),which was plausibly related to its high biomass but high salinity. In contrast, at S5, S6, and S10 relatively lower Chao1 richness but relatively higher Shannon diversity was recorded. PCoA results show that 55.12% and 21.71% of the spatial variations ofbacterial communities could be explained by the first and second axes (P<0.05), indicating a heterogeneous distribution of the bacteria in the sediments of the JZB. DOP was the most important factor loaded on the first axis, while NO3-N, NH4-N, DIN, and Chla(in the sediments) were loaded on the second axis. In terms of bacterial community structure, S5, S13, and S14 could be classified as one group, and S3, S4, S7,and S8 could be classified as another group. However,the bacterial community structure of S6 and S10 stood out clearly (Fig.2b).
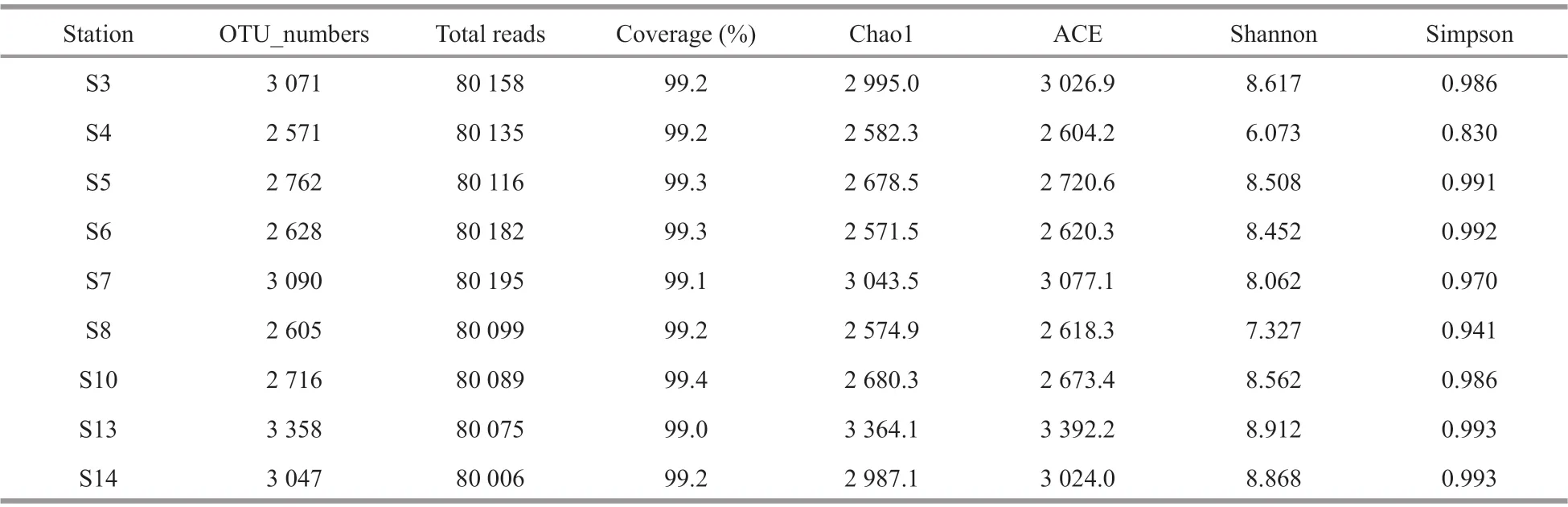
Table 4 Diversity indices at 97% sequence similarity of 16S rRNA genes calculated based on 80 117 sequences per sample of bacterial communities
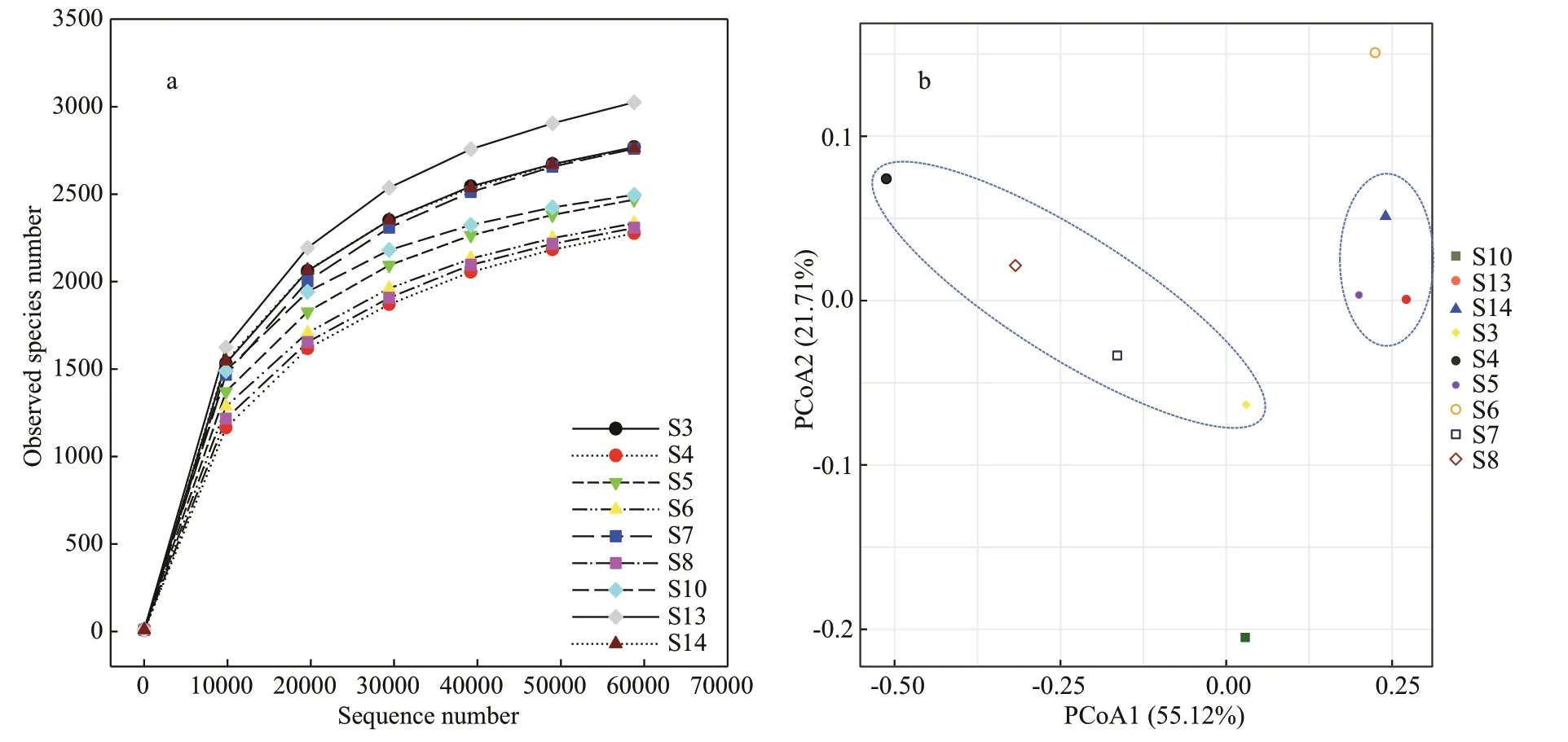
Fig.2 The rarefaction curves based on the observed species (a) and PCoA of bacterial communities in the JZB (b)
The bacterial communities across the samples were mainly composed of phyla Proteobacteria,Bacteroidetes, unidentified_Bacteria, Acidobacteria and Chloroflexi, representing 84.1% of the bacterial reads (Fig.3a). At the phylum level, the phyla Proteobacteria (64.9%–83.7%) and Bacteroidetes occupied over 80% at stations S3, S4, S5, S6, and S8, and over 75% at stations S7, S10, S13, and S14.The relative abundances of Proteobacteria and Bacteroidetes were negatively correlated with each other (Supplementary Fig.S1), indicating their opposite trophic strategies. At the class level,Gammaproteobacteria (35.4%–66.3%),Deltaproteobacteria (14.7%–26.8%), Bacteroidia and Alphaproteobacteria occupied over 74% of the total reads (Fig.3b). The relative abundance of Gammaproteobacteria had negative correlations with those of Deltaproteobacteria, Bacteroidia and Alphaproteobacteria in varying degrees(Supplementary Fig.S1). At the order level,Xanthomonadales (1.3%–46.2%), unidentified_Gammaproteobacteria (12.5%–27.3%),Alteromonadales (within Gammaproteobacteria)and Desulfobacterales (4.9%–11.7%, within Deltaproteobacteria) occupied over 30% of the total reads (Fig.3c). Negative correlations between the relative abundances of Xanthomonadales and unidentified_Gammaproteobacteria (Supplementary Fig.S1) indicated their opposite trophic strategies.
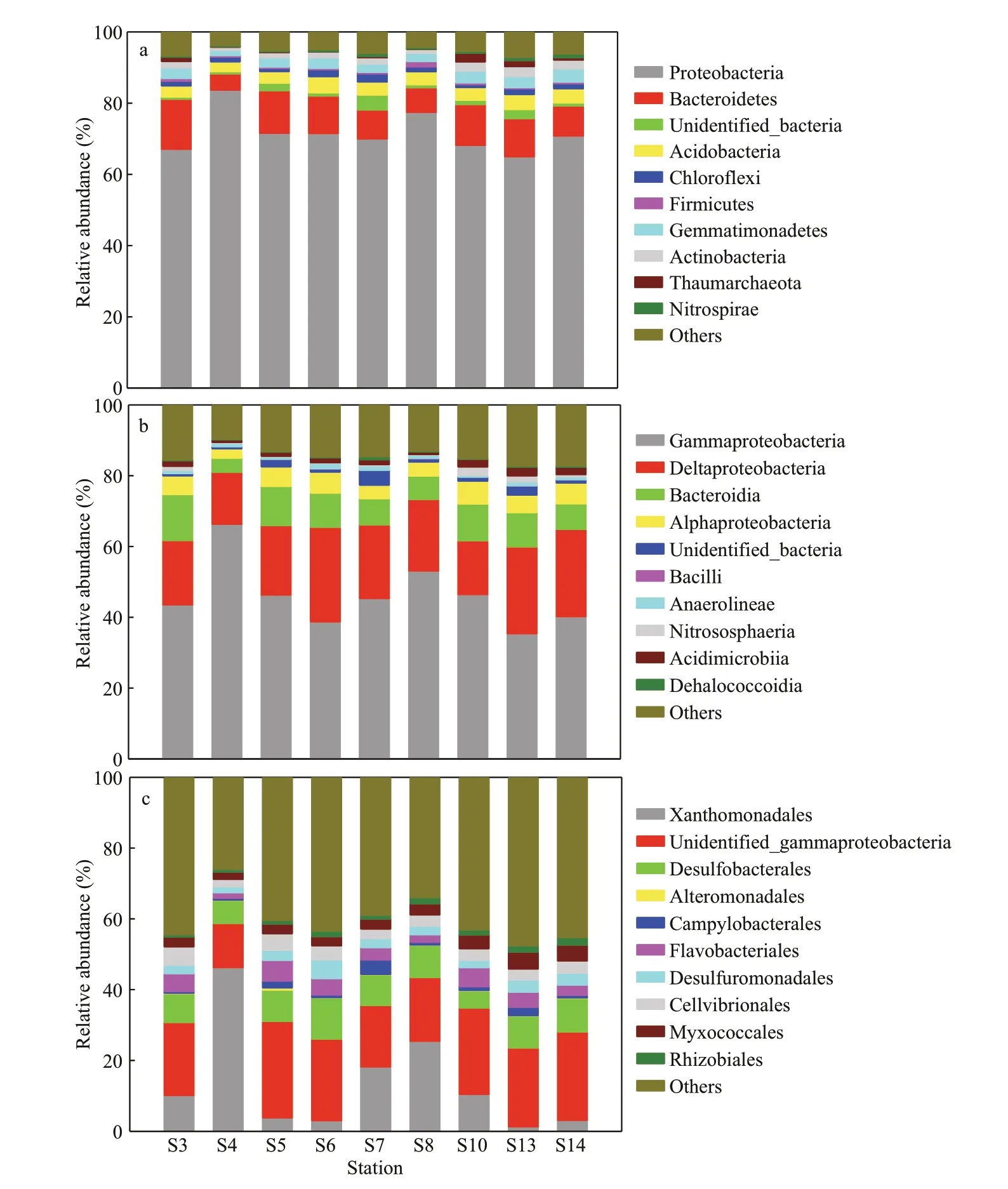
Fig.3 Relative abundances of the bacterial community composition in the sediments of the JZB at the phylum (a), class (b)and order (c) levels
3.3 Distribution patterns of bacterial communities
Higher levels of Chao1 richness and Shannon diversity indices were generally observed outside the bay such as at stations S13 and S14, and declined from the outer part to the northern part of the bay(Fig.4a & b). The relative abundance of Proteobacteria was found higher at the nearshore stations of the bay(S4: 83.7% and S8: 64.9%) and decreased from the off shore stations in the inner part to the western part of the bay and then to the outside of the bay. However,Proteobacteria tended to be abundant again away from the bay in the open sea (S14). In contrast, high levels of Bacteroidetes were observed in the southwest part of the bay and the mouth, and the levels decreased to the stations outside the bay and then to the nearshore stations S4 and S8.
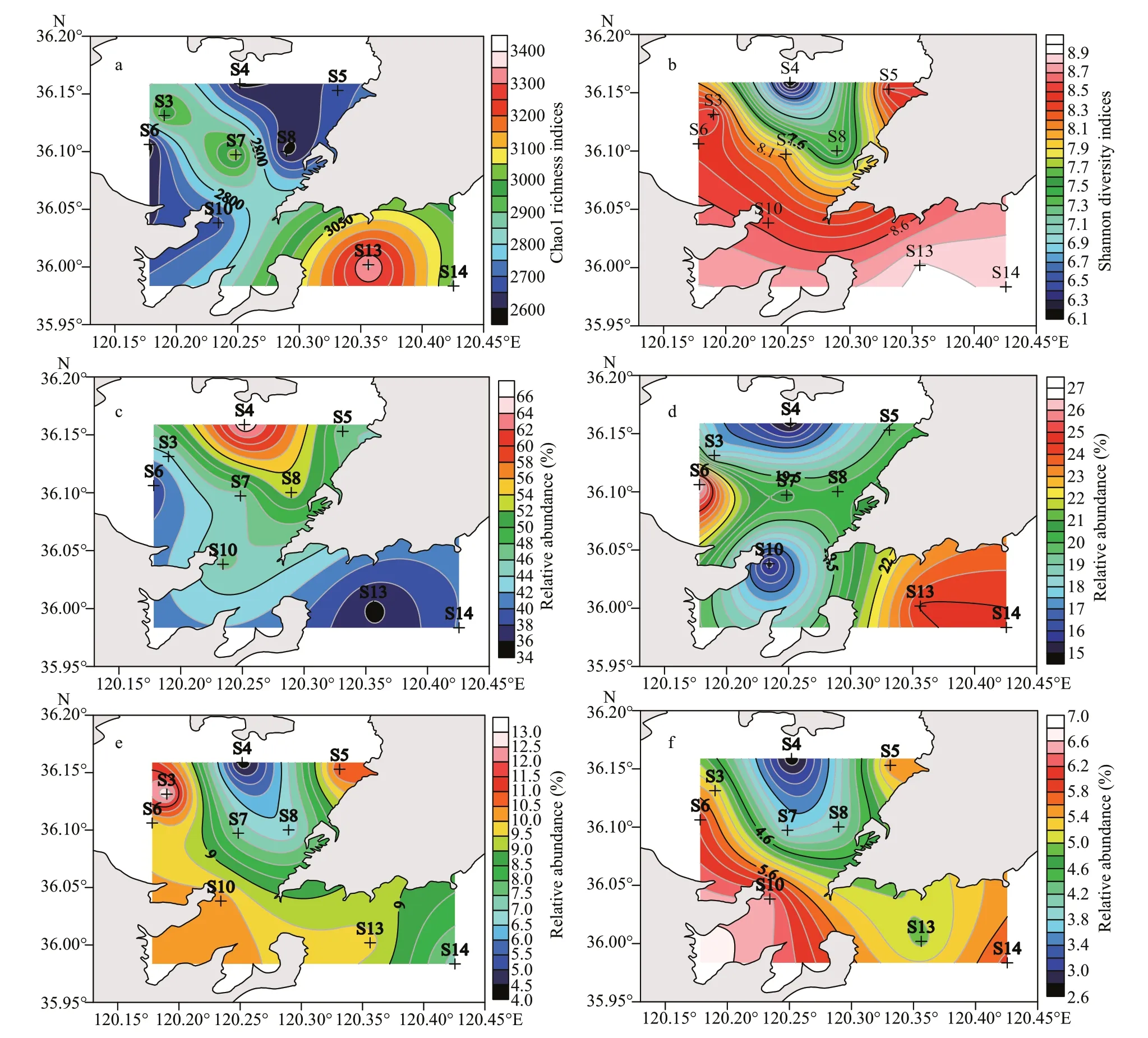
Fig.4 Spatial distributions of species richness (a: Chao1), community diversity (b: Shannon) and predominant classes (c:Gammaproteobacteria; d: Deltaproteobacteria; e: Bacteroidia; f: Alphaproteobacteria) in the sediments of the JZB
Similar to that of Proteobacteria, the relative abundance of Gammaproteobacteria was greater at the nearshore stations in the northern part of the bay(S4: 66.3%) and decreased to the mouth (S10: 46.5%),being the least in the western part of the bay (S6:38.7%) and the outside station (S13: 35.4%), which was found having better water condition in the JZB.Deltaproteobacteria generally presented an opposite pattern to that of Gammaproteobacteria, the abundance of which was higher outside the bay than at the mouth and higher in the western part than in the northern part of the bay. Similar to Bacteroidetes, the relative abundance of Bacteroidia generally increased from the nearshore stations in the northern part of the bay (S4 and S8) to the mouth and then the outer part of the bay, being the greatest in the western and eastern part of the inner bay. Alphaproteobacteria had a similar distribution pattern to that of Bacteroidia.The relative abundance of Alphaproteobacteria generally increased from the northern part of the bay to the outside of the bay and increased from the northern part of the bay to the southwest and northeast of the inner bay (Fig.4).
The relative abundance of Xanthomonadales was similar to that of Gammaproteobacteria, the value of which was the greatest at the nearshore stations (S4 and S8) and decreased from the northern part of the bay to the mouth and then to the outside of the bay. In the bay, Xanthomonadales decreased from the northern part of the bay to the west and east part of the bay. In contrast, the relative abundance of unidentified_Gammaproteobacteria increased from the northern part (S4) of the bay to the outside of the bay, being the greatest at S5. The order Alteromonadales was similar to that of unidentified_Gammaproteobacteria, being the most abundant at S5. However, the order Desulfobacterales was similar to Deltaproteobacteria, being the most abundant at S6 and the least abundant at S10.
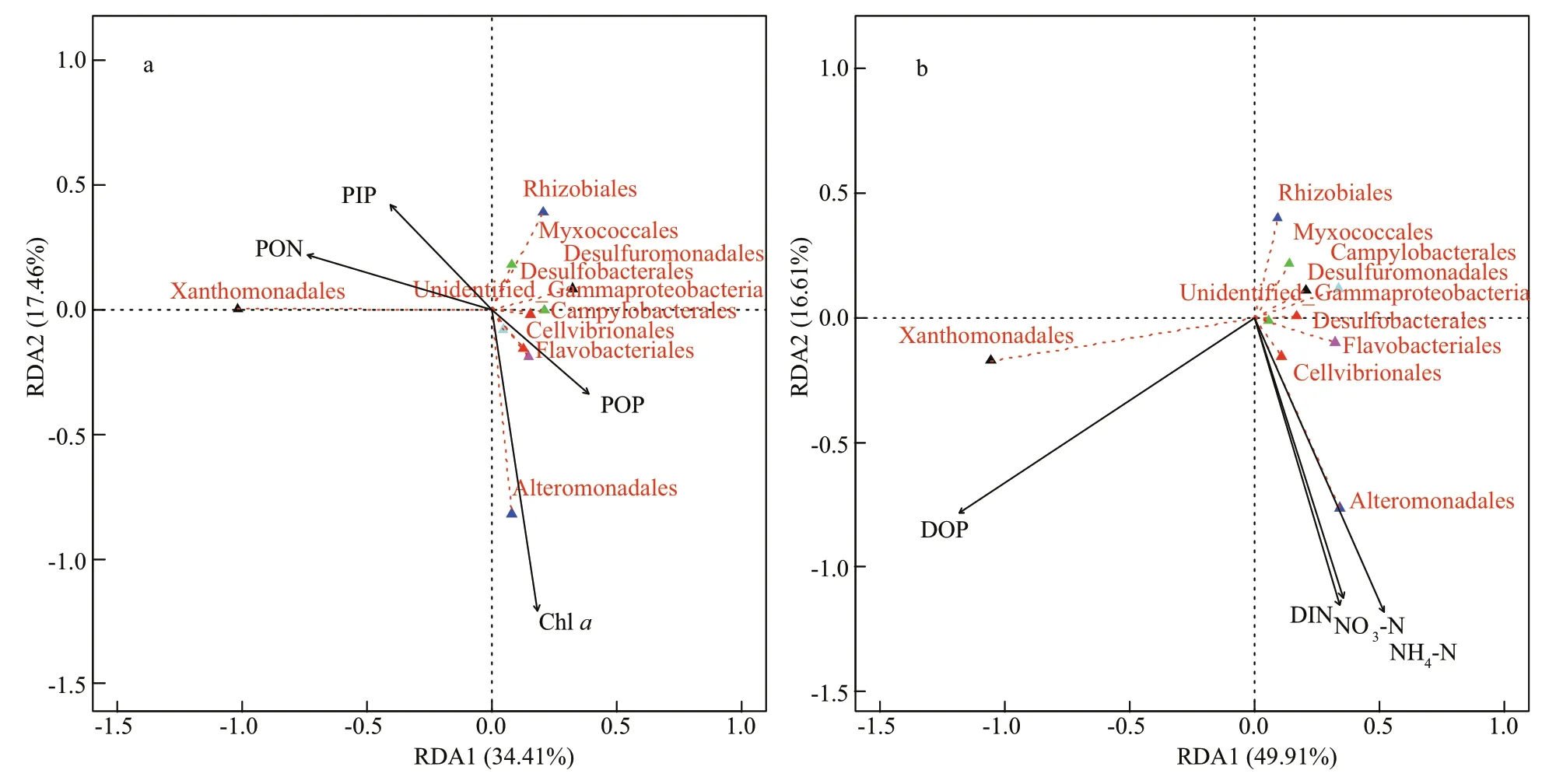
Fig.5 Ordination plots of the results from the redundancy analysis (RDA) to identify relationships between bacterial populations (red dotted arrows) and environmental variables (black solid arrows) in the sediments (a) and in the overlying seawater (b)
3.4 Linking bacterial communities to environmental variables
In the sediments, Chla, PON, PIP, and POP were selected to conduct RDA of the bacteria(Supplementary Table S3; Fig.5a), which could explain 69.4% of the bacterial community composition variation (P<0.05). 34.41% and 17.46% of the spatial variations of bacterial communities among these stations could be explained by the first two axes.However, only Chla(R2=0.65,P<0.05) was the significant influential factor on the bacterial variation,loaded on the second axis. In the overlying water,DOP, NO3-N, NH4-N, and DIN were selected to conduct RDA of the bacteria (Supplementary Table S4; Fig.5b), by which 70.6% of the variation of bacterial community composition could be explained in total (P<0.05). 49.91% and 16.61% of the variations of bacterial communities could be explained by the first two axes. In the overlying water, NH4-N (R2=0.69,P<0.05), followed by DOP (R2=0.63,P<0.1) and then DIN (R2=0.61,P=0.05) were the main factor gradient influencing the spatial variations of bacterial community composition, with only DOP loaded on the first axis. When the factors in the overlying water and in the sediments were taken together, Chla,NH4-N, NO3-N, and DOP were selected to conduct RDA of the bacteria, which could explain 70.6% of the bacterial community composition variation(P<0.05). 37.36% and 15.32% of the total variation of the bacterial communities among these stations were explained by the first two axes. DOP (R2=0.69,P<0.05), followed by NH4-N (R2=0.67,P<0.05) and then NO3-N (R2=0.56,P<0.1) were the main factor gradient influencing the spatial variations of bacterial community composition at the sediment-seawater interface (Supplementary Fig.S2).
3.5 Function prediction analysis of sediment bacteria
Correlation analysis results showed that Gammaproteobacteria were positively and Deltaproteobacteria were negatively correlated with all the OTUs referring to human disturbance and nitrogen cycling throughout the profile, which indicated the functional diversity of Proteobacteria.In addition, Deltaproteobacteria were positively correlated with the OTUs referring to respiration of sulfur compounds and sulfate respiration throughout the profile (Supplementary Table S5).
4 DISCUSSION
4.1 Bacterial distribution in the JZB and the influencing factors
The distribution of Chao1 richness and Shannon diversity indices, which generally decreased from the outside to the inner part of the bay and from the off shore to nearshore area (Fig.4), suggested that the bacterial alpha-diversity might be negatively aff ected by sediment contamination and anthropogenic pollution. The positive correlation between Chao1 richness and PINH4-N in the sediments (Supplementary Table S3) suggested that nitrogen deposition might accelerate microbial biomass and bacterial abundance through providing substrate to microbes.
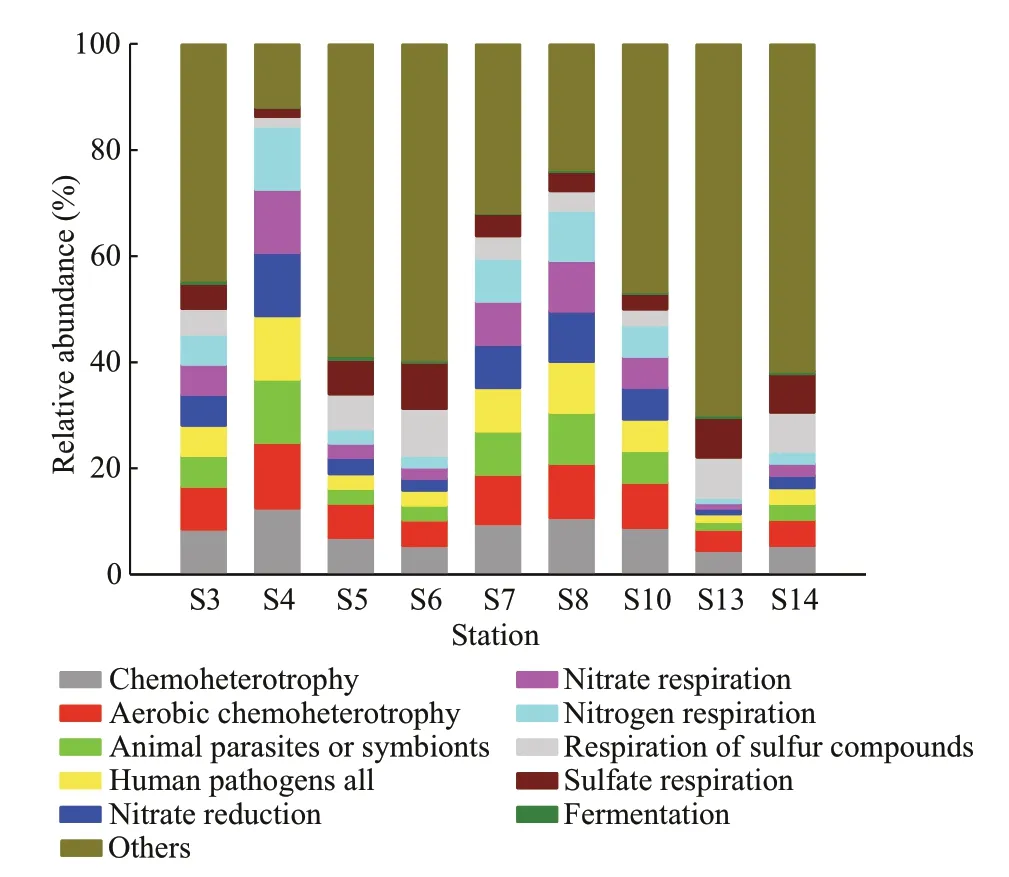
Fig.6 Relative abundances of FAPROTAX function annotation for the bacteria from the sediments in the JZB
The dominant groups in the sampled sediments diff ered from those (Proteobacteria, Acidobacteria,Bacteroidetes, Planctomycetes, and Actinobacteria)identified in the previous study at the same site (Liu et al., 2015), which plausibly suggested temporal variations ever since. Gammaproteobacteria and Deltaproteobacteria were the most abundant (Fig.3,>60%), which were consistent with typical marine sediment environment (Liu et al., 2015, 2019).Gammaproteobacteria, the most important group in marine sediments (Yu et al., 2018), were less abundant at the oligotrophic stations S6, S13 and S14, but more abundant at S4, S5, S8, and S10 with high trophic level (Fig.4), indicating their copiotrophic nature(Chigineva et al., 2009; Wang et al., 2012).Gammaproteobacteria were dominated by the aerobic hydrocarbon-degrading Xanthomonadales (Table 4),which was more abundant at S4 (46.2%) and S8(25.4%), possibly due to their roles in chemoheterotrophy, anthropogenic influence, and nitrogen cycling, while less abundant at S5, S6, S13,and S14 (<10%), due to other processes occurring there (Supplementary Table S5). RDA results showed that the negative eff ects of DOP were much more than the positive eff ects of NH4-N and NO3-N on Xanthomonadales (Fig.5b). Despite lower abundance,the subgroup Alteromonadales was the most active Gammaproteobacteria, indicating its importance in nutrient cycling and overall ecosystem functioning.The positive correlations between the order Alteromonadales and all the dissolved inorganic nutrients (Supplementary Table S4) indicated their copiotrophic nature. In addition, positive correlations of Alteromonadales with Chlaboth in the sediments(Supplementary Table S3) and in the overlying water(Supplementary Table S4) suggested their relevance in biomass construction, as Alteromonadales was identified as the only group with genes involved in aerobic anoxygenic photosynthesis (Cleary et al.,2017). Therefore, the most abundant Alteromonadales at S5 across stations was the joint results of the greatest nutrients (nitrogen) in the seawater and highest Chladerived from the sediments (Tables 1 &2). RDA results (Fig.5) show that DOP had positive eff ects on Alteromonadales such as Chla, NH4-N, and NO3-N, and the former might have been transferred into DIP after being utilized by these bacteria. The order Cellvibrionales (within Gammaproteobacteria)was negatively correlated with PIP in the sediments,indicating that PIP was disadvantageous to the survival of this order and sediments with more PIP tended to have less abundant Cellvibrionales andviceversa. So far, Cellvibrionales in the JZB or its adjacent Yellow Sea has been seldom reported, despite its role in P cycling. RDA results showed that Chla(in the sediments) had greater eff ects on bacterial communities than PON, POP, and PIP did in the sediments (Fig.5a). Overall, DOP had important eff ects on the most active orders Xanthomonadales and Alteromonadales, and became the most important factor shaping the distribution pattern of sediment bacteria in the JZB (Supplementary Fig.S2). This was consistent with the habitat status of phosphate insuffi ciency in the bay (Yuan et al., 2018). These results supported the idea that sediment bacteria are profoundly influenced by their habitat in the overlying seawater.
Contrasting distribution patterns of Deltaproteobacteria (i.e., the most abundant at S6,S13, and S14 but the least abundant at S4 and S10,Fig.4) and Gammaproteobacteria indicated the oligotrophic nature of Deltaproteobacteria, which contrasted with the previous report in which Deltaproteobacteria was found abundant in entrophic and anoxic sediments (Bowman and McCuaig, 2003;Liu et al., 2011). The patterns of the predominant members Desulfobacterales and Desulfuromonadales(more abundant at S6, S13, and S14 but less abundant at S4 and S10) were similar to Deltaproteobacteria, as they are chemoorganotrophs related to the degradation of recalcitrant organic matter in global marine sediments (Plugge et al., 2011). It is notable that these predominant orders were in perfect trade-off at S4,where the most abundant Xanthomonadales was accompanied by the least abundant unidentified_Gammaproteobacteria. Considering the major eff ects of DOP on Xanthomonadales, we proposed that the riverine inputs at S4 might contain the organic pollutants. The distribution pattern of Alphaproteobacteria was also nearly opposite to that of Gammaproteobacteria, which was less abundant at S4, S7, and S8 but more abundant at S10, S6, and S14(Fig.4). Alphaproteobacteria was the most abundant at S10 across stations, which suggested that the subgroup was obviously stimulated by the crude oil refining plants and/or played a crucial role in oildegrading. The predominant order Rhizobiales could play a critical role in carbon and nitrogen cycling of marine ecosystems through photosynthesis. Both orders Myxococcales and Rhizobiales presented a decreasing trend with the increasing TOC in the sampling stations (from S3 to S14) in the overlying water (Supplementary Fig.S1), possibly due to their susceptibility to the more recalcitrant substrates(Cleary et al., 2017). However, such process was depressed by DOP (Supplementary Table S4). The class Bacteroidia was more abundant at the nearshore stations S5 and S10 while less abundant at the nearshore stations S4 and S8 (Fig.3), which might suggest diff erent substrate types from riverine inputs.The predominant order Flavobacteriaceae within Bacteroidia could utilize all kinds of substrates but was extremely enriched with the occurrence of starch(Kieft et al., 2018). Therefore, these bacteria may be enriched because of deposition of the recalcitrant organic matter from the riverine inputs at S5 and S10.
With regard to some rare phyla, unidentified_Bacteria were the most abundant at S7 across stations(Fig.3) possibly due to PIN and PON being the greatest there (Table 2), which was confirmed by the positive correlations between them (Supplementary Table S3). In this study, unidentified_Bacteria were presumed to be the class Epslionproteobacteria and the predominant order Campylobacterales were reported to be responsible for most denitrification genes in the anaerobic layers of oceans (Llorens-Marès et al., 2015). Therefore, it was considered that S7 (in the deep-water area at the center of the bay)might have anaerobic conditions that were advantageous for the survival and reproduction of these taxa. Similar abundant Campylobacterales at S5 and S13 might also indicate anaerobic conditions.Considering the consistent ChlaandSin the overlying water, the greatest Campylobacterales at S7 among stations was also related to the unique condition characterized by high salinity and high biomass. As such, phyla Chloroflexi and Nitrospirae that were closely related to Chlaalso hiked at S7. The distribution of Acidobacteria (more abundant at S6,S13, and S14 but less abundant at S3, S4, and S5) was possibly related to their susceptibility to the more recalcitrant substrates, i.e., oligotrophic nature (Fierer et al., 2007; Zhang et al., 2016). The negative correlation between Acidobacteria and DOP suggested their involvement in P cycling (Supplementary Table S4). In addition, the ubiquitous Acidobacteria in the JZB might also imply acidification.
4.2 Possible bioindicator taxa in the assessment of the anthropogenic pollution
Our results showed that anthropogenic pollution profoundly altered the bacterial community composition among stations, which could be indicated by the order Xanthomonadales (Gammaproteobacteria)(Fig.3c). That was, S4 (near the Hongdao aquiculture area) and S8 (near the Qingdao Port), harbored the most abundant Xanthomonadales due to the most river input and livestock wastes. S7 (located at the center of the bay), S10 (at the mouth), and S3 (at the northwest) had comparable Xanthomonadales and unidentified_Gammaproteobacteria. However, both S5 (located at the estuary of Licun River) and the lesspolluted S6, S13, and S14 held the most abundant unidentified_Gammaproteobacteria. Overall, the bacterial community composition transitioned from being Xanthomonadales-dominant at S4 and S8 to being unidentified_Gammaproteobacteria-dominant at S5, S6, S13, and S14. The predicting function correspondingly changed from being nitrogen cycling-dominant at S4 and S8 to being sulfur cyclingdominant at S5, S6, S13 and S14 (Supplementary Table S5). This could be partly caused by the organic pollutants from livestock and industrial wastes that entered the JZB. The order Xanthomonadales (genusStenotrophomonas) has been used as a bioindicator for the detection of the impact of multiple contamination sources by polycyclic aromatic hydrocarbons (PAHs) on the bacterial community(Juhasz et al., 2000; Kanaly and Harayama, 2010).Strains of the genusStenotrophomonashave been reported as PAHs- and Triton X-100- degrading bacteria (Chen et al., 2004; Gutierrez et al., 2013).Therefore, we proposed that the order Xanthomonadales holds the potential ofindicating the anthropogenic organic pollution in the JZB. On the other hand, there were positive correlations between Alteromonadales and dissolved inorganic nutrients rather than organic components(Supplementary Table S5), which indicated their sensitivity to the dissolved inorganic nutrients and suggested the potential of this subgroup as a bioindicator ofinorganic pollution. As reported,Alteromonadales are mainly aerobic chemoheterotrophic species that decompose the primary produced carbon sources (Naghoni et al.,2017) and accounted significantly for the DICassimilating Gammaproteobacteria (DeLorenzo et al.,2012), which might be enhanced by the inorganic nutrients. However, inorganic pollution possibly failed to change the composition of the bacterial communities in the JZB, which is why the bacterial community composition at S5 showed a little diff erence from those of S13 and S14 in the open sea.In addition, the positive correlation between Deltaproteobacteria and the OTUs referring to the respiration of sulfur compounds and sulfate respiration(Supplementary Table S5) verified the application of Deltaproteobacteria as a sensitive indicator of sulfatereducing bacteria (SRB) (Ravenschlag et al., 2000;Liu et al., 2015). Deltaproteobacteria could also serve as a negative indicator of anthropogenic pollution in the JZB due to their negative correlation with all the OTUs referring to anthropogenic influences and nitrogen cycling.
In this study, the Pearson correlations between bacterial orders and environmental factors were confirmed by the redundancy analysis, withP<0.05 being set as the significant level (n=9), while those between bacterial orders and OTUs indicating biogeochemical processes were confirmed by literature, both excluded the causality of correlation results. Our results provided profound evidence supporting the indicating function of bacteria.Nevertheless, given the extreme complexity of microbial communities in marine ecosystems in terms of distribution and functions, our results should be interpreted with great caution regarding the indicating functions of bacteria, which needs further experimental verification and evidence. This study supports the idea that bacterial communities could serve as sensitive indicators of anthropogenic pollution,providing an eff ective and sensitive tool for rapid environmental assessment (Simonin et al., 2019).
5 CONCLUSION
The bacterial communities in the sediments presented considerable spatial variations across the JZB, with Proteobacteria and Bacteroidetes being the most abundant. More specifically, bacterial alphadiversity indices decreased from the outside to the inner part of the bay and from the off shore to the nearshore area. Bacterial community structures of S3,S4, S7, and S8 clustered; those of S5, S13, and S14 could also be classified as one group, while those of S6 and S10 were distinct from each other and from the other stations. Overall, the bacterial community composition transitioned from being Xanthomonadales-dominant at S4 and S8 to being unidentified_Gammaproteobacteria-dominant at S5,S6, S13, and S14, while in other stations they had comparable values. Among stations,Gammaproteobacteria (order Xanthomonadales)were more abundant at the nearshore stations (S4 and S8) which receive the most freshwater inputs and suff er from the livestock wastes but were less abundant in stations having better environmental conditions (S5, S6, S13, and S14). The distribution,together with their positive correlations with the OTUs referring to human activities and nitrogen cycling, indicated the potential of Xanthomonadales as a sensitive indicator for sediment contamination and/or anthropogenic pollution. Deltaprotepbacteria presented opposite distribution patterns with Gammaproteobacteria, suggesting their contrasting trophic strategies (oligotrophic vs. copiotrophic). The bacterial distribution was aff ected by especially the factors (DOP) in the overlying seawater, possibly related to the habitat status of a general P insuffi ciency in the bay.
Since microbes responding to the overweight inputs of nutrients are indicative for the estimation of anthropogenic influence on the biogeochemical processes, this work is of great importance for understanding coastal ecosystems and human influence on them. Our results identified the relevance of bacterial information in nutrient cycling as well their indicating functions to sediment contamination and/or anthropogenic pollution. Bacterial indicators could complement the information regarding humaninduced impacts, which however has not been well researched. Further investigation of specific bacterial taxa responding to selected pollutants may help to improve our understanding of the indicating functions of bacterial communities.
6 DATA AVAILABILITY STATEMENT
The datasets generated during the current study are available from the corresponding author on reasonable request.
7 ACKNOWLEDGMENT
The authors thank the crews of R/VChuangxinfor supports in data collection, and Prof. SUN Xiaoxia for support in Chladata processing.
杂志排行

Journal of Oceanology and Limnology的其它文章
- Steady increase in water clarity in Jiaozhou Bay in the Yellow Sea from 2000 to 2018: Observations from MODIS*
- Phylogenetic diversity and bioactivity of culturable deepsea-derived fungi from Okinawa Trough*
- Allelopathic eff ects of mixotrophic dinoflagellate Akashiwo sanguinea on co-occurring phytoplankton: the significance of nutritional ecology*
- Investigation of the decline of Ulva prolifera in the Subei Shoal and Qingdao based on physiological changes*
- Effi ciency of phosphorus accumulation by plankton,periphyton developed on submerged artificial substrata and metaphyton: in-situ observation in two shallow ponds*
- Petroleum exploitation enriches the sulfonamide resistance gene sul2 in off shore sediments