膜性肾病关键基因的生物信息学分析
2021-06-13窦涪琳刘庆珍徐巧莹刘海英傅余芹
窦涪琳,刘庆珍,徐巧莹,刘海英,傅余芹
山东大学第二医院,济南 250021
膜性肾病(MN)是成人肾病综合征最常见的病理类型,其病理特征为弥漫性肾小球基底膜增厚伴上皮下免疫复合物沉积。MN是由自身免疫反应引起的肾小球损害。自身抗体与足细胞靶向结合引起了免疫复合物沉积、肾小球基底膜增厚以及足细胞形态改变,从而导致蛋白尿的进展[1]。近年来在MN中发现了多种自身抗原,包括分泌型磷脂酶A2受体(PLA2R1)[2]、含7A的血栓反应蛋白1型结构域(THSD7A)[3]和神经表皮生长因子样1蛋白(NELL-1)[4],分别占MN患者的70%~80%、3%~5%及5%~10%。外周循环抗体的测定已成为临床重要的参考指标。体内外实验证据表明多种通路和细胞因子在MN的发病机制中起关键作用。然而,MN的分子机制尚不完全清楚。生物信息学分析允许在基因组水平上进行基因改变的筛选。然而,独立的微阵列分析常导致假阳性率。本研究从基因表达公共数据库(GEO)中获得MN患者的两个微阵列数据集(GSE108109、GSE115857),将原始数据进行一系列处理后,识别MN相关的Hub基因,应用NephonSeq v5平台分析Hub基因与MN临床特征的相关性,以便进一步探讨MN的发病机制以及涉及的关键基因。
1 材料与方法
1.1 微阵列数据搜索和下载 本研究中的微阵列数据从基因表达数据库(GEO)下载(https://www.ncbi.nlm.nih.gov/geo)。搜索的关键词为“膜性肾病”,且符合以下标准:研究类型为表达谱基因芯片;组织来源为人类肾组织。基于上述标准,选择了GSE108109、GSE115857这两个数据集。GSE108109基于GPL19983,包括MN组44例,正常对照组6例;GSE115857基于GPL14951,包括MN组11例,正常对照组7例。
1.2 差异表达基因(DEGs)识别 用GEO2R网络工具(http://www.ncbi.nlm.nih.gov/geo/geo2r)分别对GSE108109、GSE115857进行分析,得到MN组与正常对照组的差异基因ID,其中筛选条件为|log2FC(差异倍数)|>1且校正后的P值(adj.P)<0.05。利用平台信息文件将表达谱芯片中的探针矩阵转化为基因矩阵。若没有相应的基因符号就剔除,若有多个基因探针组,则在探针组中取中位数。随后,将上述两组差异基因上传到Venn图中,重叠基因即为本研究中的DEGs。
1.3 DEGs的基因本体论(GO)和京都基因与基因组百科全书(KEGG)富集分析 将DEGs上传到DAVID在线数据分析工具,进行GO富集和KEGG富集分析,其中P<0.05表明在统计学上差异有统计学意义(6.8版,http://david.ncifcrf.gov)。
1.4 蛋白质—蛋白质相互作用(PPI)网络构建与Hub基因识别 用STRING工具(10.0版本,http://string-db.org)构建DEGs的PPI网络,探索DEGs与互作网络之间的联系。其中综合得分阈值>0.4表明在统计学上有显著的相互作用。用Cytoscape软件(6.3版,http://www.cytoscape.org/)来进一步分析和可视化PPI网络数据。应用CytoHubba中的“Degree”分析方法,从所有DEGs中发现特征节点并识别Hub基因,其中Degree≥7的基因为Hub基因。
1.5 Hub基因分析 应用NephonSeq v5在线平台(http://v5.nephroseq.org),对MN患者Hub基因的表达与血肌酐、蛋白尿之间进行皮尔逊相关性分析,|r|>0.5表示两者有相关性,|r|值越大表示相关性越好,正数代表正相关,负数代表负相关,P<0.05为差异有统计学意义。
2 结果
2.1 MN中的DEGs GSE108109、GSE115857中分别有1 478和4 345个差异基因,有240个DEGs,DEGs中分别有149个上调基因和91个下调基因(图1)。
2.2 MN中DEGs的GO与KEGG分析结果 MN中DEGs的GO分析结果显示,其生物学过程(BP)显著富集在细胞形态调节、肌动蛋白细胞骨架组织的调节、脂蛋白代谢及B细胞稳态等过程,其细胞成分(CC)显著富集在质膜、基底膜、细胞间桥等,其分子功能(MF)显著富集在氧化还原酶活性、蛋白激酶C及细胞周期蛋白结合等;KEGG分析显示,DEGs显著富集在p53信号通路、脂肪细胞脂解的调节及凋亡等。见图2。
2.3 DEGs的PPI网络及Hub基因 DEGs的PPI网络(图1)平均节点连通度为3.11,平均局部聚类系数为0.156,蛋白相互连接数为224。Hub基因包括TP53、PIK3R1、KIT、QSOX1、LAMB1、SHC1、CDK2、APOL1、NES、PTPN6、POLR2A、PRKACA、LTBP1基因。在PPI网络中,Hub基因与其他基因联系紧密(图3)。13个Hub基因中,10个基因表达上调,包括TP53、QSOX1、SHC1、CDK2、APOL1、NES、PTPN6、POLR2A、PRKACA、LTBP1,3个基因表达下调,包括LAMB1、PIK3R1、KIT。

注:椭圆形节点代表上调基因,菱形节点代表下调基因。

图2 MN中DEGs的GO和KEGG富集分析
2.4 Hub基因与MN临床特征的关系 MN患者肾组织中,PRKACA、LTBP1基因表达与蛋白尿呈正相关(r分别为0.858、0.799,P分别为0.006、0.017),POLR2A的表达与蛋白尿呈负相关(r=-0.866,P=0.005)。另外,PRKACA基因表达与血肌酐呈负相关(r=-0.601,P=0.008),POLR2A的表达与血肌酐呈正相关(r=0.583,P=0.011)。
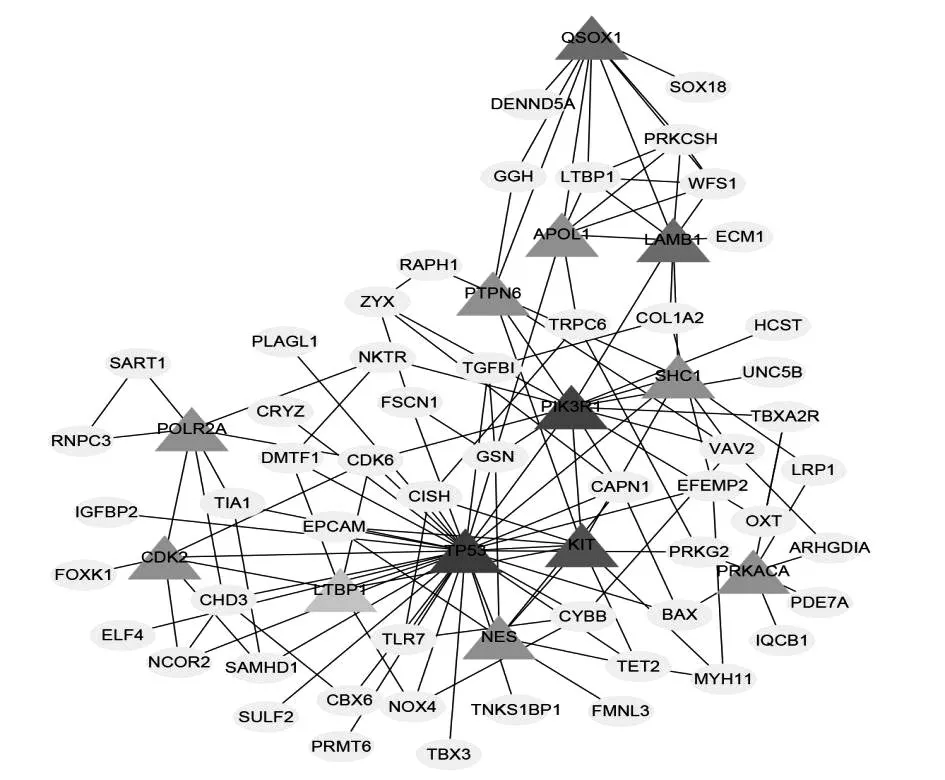
图3 Hub基因的PPI网络
3 讨论
MN是导致亚洲成年人肾病综合征的最常见原因之一,约占30%。MN患者的临床病程差异悬殊,约40%的患者会自发缓解,而30%的患者对免疫抑制治疗效果不佳,最终进展至终末期肾病[5]。因此对MN的免疫抑制治疗开始时机及治疗力度一直存在很大的争议,MN患者的精准化治疗也由此应运而生,而治疗的关键是识别疾病的确切病因及发病机制。在不到十年的时间里,MN分子病理机制的研究发展迅速,有些已广泛应用于临床,但目前仍有很多潜在未知的机制存在。本研究应用生物信息学方法探讨MN的分子机制,并试图寻找潜在的新治疗靶点。共筛选出240个差异基因,它们参与细胞形态调节、细胞骨架构建等生物学功能。其中13个基因被认定是Hub基因,包括TP53、PIK3R1、KIT、QSOX1、LAMB1、SHC1、CDK2、APOL1、NES、PTPN6、POLR2A、PRKACA、LTBP1。
TP53/P53,参与细胞周期与细胞凋亡的调节,与多种人类肿瘤有关,其中包括肾透明细胞癌。曾有研究报道,iNOS和P53在IgAN固有肾细胞中表达上调,可能共同诱导细胞凋亡活性,从而介导IgA肾病进行性肾损伤[6]。微阵列分析表明TP53编码的蛋白与肾功能衰竭的进展有关[7]。LTBP1基因编码的蛋白,属于潜在转化生长因子-β结合蛋白家族(LTBPs),它是TGF-β分泌与激活的调节蛋白之一。HUANG等[8]发现LTBP-1可能通过调节TGF-β活性参与糖尿病肾病的发病过程。LEE等[9]报道,MN患者足细胞中TGF-β的表达上调。TGF-β通过多个方面导致慢性肾脏病的肾纤维化过程,包括近端肾小管功能障碍、上皮细胞修复和再分化失败,以及随之而来的肾小管间质纤维化[10]。最近的研究发现,TP53是TGF-β诱导的信号通路中的一个辅助因子,并且是几种TGF-β促纤维化反应基因的转录辅助调节因子,以TP53为靶点的药理学和遗传学方法减弱了相关基因的表达,减轻了肾纤维化反应,证实了TP53在肾脏疾病中的关键作用[11]。结果显示,在MN患者的肾脏组织中,LTBP1与蛋白尿呈正相关,提示LTBP1参与MN患者肾损伤的进展,可能与激活TGF-β有关,并且协同TP53参与了TGF-β1诱导的肾损伤过程。CDK2编码的蛋白参与细胞周期调控,微阵列分析表明CDK2编码的蛋白与肾功能衰竭的进展有关[7]。抑制新月体肾炎小鼠的CDK2活性,可抑制足细胞增殖从而改善肾功能[12],提示抑制CDK2活性是治疗以足细胞增殖为特征的肾小球疾病的潜在靶点。而SAURUS等[13]发现,阻止CDK2表达下调,可保护足细胞免于凋亡。提示CDK2基因的异常表达,无论是高表达还是低表达,均会对足细胞产生影响。
SHC1在多种组织中表达,包括肾细胞,参与调节细胞氧化应激的应答。MARTIN等[14]发现SHC1在蛋白尿性肾病中过度表达,SHC1的增加导致足细胞裂孔隔膜的关键结构成分变形和分解,破坏了滤过屏障的完整性。p66Shc是Shc1基因编码的三种衔接蛋白之一,p66Shc作为一种氧化还原酶,导致氧减少、ROS产生增多,引起线粒体肿胀,并导致CytC释放到胞质中,迫使细胞沿凋亡途径下行[15]。p66Shc与多种肾脏疾病的进展及恶化相关[16],该基因在糖尿病肾病患者肾组织中表达上调,且被认为是肾小管氧化损伤的潜在生物标志物[17-18]。MILLER等[19]报道,p66Shc参与调节肾血管张力,在高血压肾病大鼠中过度表达,促使高血压肾病肾血管功能受损。此外,该基因还参与年龄相关性肾脏病变[20]、抗癌药物肾毒性[16]的进展。据报道,APOL1高风险变异体(G1和G2)与非洲血统慢性肾脏病的高发病率相关,被认为是肾风险变异。由于足细胞有限的修复和再生能力,并且对自噬相对敏感,所以足细胞容易受到载脂蛋白1野生型(G0)和高风险变异体(G1和G2)的影响[21]。另外,APOL1基因也被全基因组关联研究鉴定为足细胞病的易感基因之一[22]。另外,APOL1的表达可能会在肾脏积聚并导致炎症加剧[22]。APOL1高风险变异体引起的肾脏病理表现有局灶节段性肾小球硬化、塌陷性肾小球疾病和伴有动脉肾硬化的非特异性局灶球性肾小球硬化,而且APOL1高风险变异体可影响多种疾病,如膜性肾病[23]、狼疮性肾炎、糖尿病肾病的预后,此外,PLA2R抗体相关IMN患者中,APOL1高风险变异体的肾脏病理表现为塌陷性肾小球疾病[23]。
PRKACA基因编码的蛋白是参与细胞凋亡的蛋白激酶A亚单位之一。目前,PRKACA扩增是鉴定库欣综合征相关基因缺陷的一种方法。先前的研究通过微阵列分析观察到,IgA肾病患者单核细胞会出现该基因的表达改变,可能通过调节IgAN进程中的凋亡来发挥其在单核细胞中的作用[24]。本结果显示在MN患者中,PRKACA的表达与血肌酐呈负相关,与蛋白尿呈正相关,提示PRKACA基因的异常表达,均会对疾病进展产生影响。PTPN6是蛋白酪氨酸磷酸酶(PTPs)家族的一员。PTPs被认为对损伤和再生反应具有多方面的调节作用。有多项研究发现,糖尿病大鼠的PTPN6表达增加,通过引起VEGF抵抗,降低足细胞中neparin的磷酸化水平,从而导致足细胞凋亡[25]。而近期研究表明,肾缺血再灌注大鼠PTPN6基因敲除后,肾小管上皮细胞促凋亡信号被过度激活[26]。应用生物信息学方法检测慢性肾小球肾炎大鼠PIK3R1基因的表达,发现该基因的表达明显升高,可能是慢性肾小球肾炎发病的关键基因[27]。在一项荟萃分析中,PIK3R1被鉴定为有肾脏功能的基因[28]。
到目前为止,很少有研究提到KIT、QSOX1、NES、LAMB1和POLR2A与MN的相互作用关系。KIT,通常被称为原癌基因c-kit,编码一种酪氨酸激酶受体,可磷酸化多种细胞内蛋白,这些蛋白在多种细胞类型的增殖、分化、迁移和凋亡中发挥作用。QSOX1在多种肿瘤疾病中过度表达,尤其是乳腺癌。NES,该基因主要在神经细胞中表达,相关的疾病包括室管膜母细胞瘤和脑室周围白质软化症。LAMB1,是基底膜的主要非胶原成分,与多种生物学过程有关,包括细胞粘附、分化、迁移、信号传导、突起生长和转移等。在MN患者中,POLR2A的表达与血肌酐呈正相关,与蛋白尿呈负相关,推测POLR2A的异常表达与MN患者肾损伤的进展密切相关。
尽管本研究中部分潜在的Hub基因在既往MN的相关文献中未被报道,但这些基因在足细胞损伤、肾纤维化等病理过程中的作用已被大量实验证实,提示它们可能在MN的发生和发展中也起重要作用。然而,本研究存在一定的局限性。首先,来自GEO的临床资料并不适用于每个样本。此外,微阵列数据来自MN的不同阶段,不同阶段的某些基因表达水平可能不完全相同。最后,这些潜在候选基因的具体分子机制和生物学功能仍有待进一步实验研究验证。本研究通过生物信息学分析肾组织微阵列数据,发现240个DEGs和13个新的潜在的Hub基因,这些基因可能是MN潜在治疗靶点的生物标记物。这为加深MN分子机制的理解提供了科学可靠的数据,为今后MN研究提供了宝贵资源。
