普通小麦Soru#1×Naxos群体代谢物的QTL定位
2021-06-08薛明云陈杰陈伟吴纪中兰彩霞李东芹
薛明云陈杰陈伟吴纪中兰彩霞李东芹
1.华中农业大学植物科学技术学院,武汉 430070; 2.华中农业大学作物遗传改良国家重点实验室,武汉 430070;3.江苏省农业科学院种质资源与生物技术研究所,南京 210014
代谢物在植物正常生长发育以及适应不同环境条件过程中扮演着不可或缺的角色[1-2]。与此同时,这些代谢物也是人类所需营养元素的重要组成部分[2-4]。例如,具有不同化学修饰类别的黄酮类物质与水稻或者拟南芥在地球不同纬度分布状况相关联[5-6],而人类饮食结构中的这类黄酮代谢物具有良好的抗炎症等生物活性[7-8]。由于代谢物种类数量庞大[9-10]且变异丰富[11],通过结合代谢组学检测手段与不同的遗传学设计,能够帮助我们更系统地研究代谢物在植物体内的含量变化及其可能的生物学意义。该研究思路已经成功应用于水稻、番茄和玉米等多种作物的代谢组学研究中[12-14],相关研究结果有助于解析代谢物与植物响应逆境[6,15]或者与食物口感风味[16-17]等之间的关联。例如,代谢组学已经成功用于解析番茄风味和果皮颜色的遗传基础和代谢机制[14],为番茄果色和风味育种提供了理论指导。玉米中的mQTL分析[13]则有助于帮助我们全面了解玉米初生代谢物调控网络。
然而,与其他作物相比,这种将代谢物与数量性状位点(quantitative trait loci)相结合(mQTL)或者与全基因组关联分析(genome-wide association study)相结合(mGWAS)的方法在小麦研究中仅有少量报道。在这些研究中,早期的mQTL研究由于没有参考基因组信息,未能提供任何候选基因[18-19];随着小麦基因组测序工作的进行,之后的小麦mGWAS结果能够提供可能的候选基因信息[20],但是未进行后续验证。目前,在小麦中已经能够使用酶活功能验证等方式对小麦mGWAS位点候选基因的功能进行初步验证[21],还能尝试探讨这些代谢物与小麦农艺性状之间的可能关联[22]。与水稻、玉米等作物相比,小麦代谢组学相关研究,尤其是将代谢组学测定方法与遗传学设计相结合的研究方式现在还处于起步阶段,需要进一步研究,一方面促进对控制小麦不同代谢物的潜在遗传基础的了解,另一方面则有助于揭示复杂农艺性状背后的分子机制,从而为小麦遗传改良提供理论基础和分子资源。
本研究使用由双亲Soru#1和Naxos构建的重组自交系(RIL)群体材料。由于双亲的抗性差异,前人已利用该RIL群体进行了小麦赤霉病和条锈病等的QTL定位研究,获得了潜在抗性数量性状位点[23-24]。然而,目前还没有针对该群体代谢组学的研究报道。通过对其进行代谢组学检测,能够了解群体中不同材料的籽粒代谢谱。与此同时,结合代谢谱信息和对应材料基因型信息进行mQTL定位,能够获得可能调控不同代谢物含量的数量性状位点,为候选基因克隆和功能验证等后续研究提供基础数据。另外,还可以将代谢物QTL和抗病QTL信息进行联合分析,挖掘可能同时影响特定代谢物含量与病害抗性的位点,从而为小麦抗病育种研究提供新的视角。
1 材料与方法
1.1 试验材料
本研究使用由Soru#1和Naxos杂交所形成的含有119个株系的F2∶9代RIL群体,其中Soru#1为国际玉米小麦改良中心(CIMMYT)育种的1个六倍体人工小麦品系[23],系谱为SABUF/5/BCN/4/RABI//GS/CRA/3/AE.SQUARROSA(190)。Naxos为德国小麦品种,系谱为Tordo/St.Mir808-Bastion//Miranet。该群体成熟籽粒由江苏省农业科学院吴纪中研究员团队提供。对大田收获的成熟籽粒随机取5粒进行研磨,即为1个生物学重复。
1.2 代谢物提取与检测
晒干后的种子使用植物组织研磨仪(Qiagen,Germany)于29 Hz研磨1 min后,按照1 000 μL/0.1 g粉末的比例加入70%甲醇用于提取小麦籽粒代谢物。代谢物提取过程在冰上进行,提取液(70%甲醇,预先加入阿昔洛韦至终质量浓度0.1 mg/L作为内标)与粉末充分混合后,每10 min涡旋混匀1次,涡旋3次后将混合溶液于4 °C过夜浸提,于10 000 r/min离心5 min后,取上清经0.22 μm孔径滤膜(上海安谱)过滤后,即为代谢物样品。代谢物检测按照参考文献[25]方法使用LC-ESI-MS/MS系统进行。对于检测到的代谢物信号,使用步进多反应监测(sMRM)对每种代谢物在不同小麦籽粒样品中的相对含量进行定量检测[26]。每个sMRM检测窗口为90 s,检测时长1 s,使用Analyst 1.5软件进行数据分析。由于代谢物在不同生物学样品之间的含量变异远远大于技术重复之间的变异[21],本次代谢物检测未进行技术重复。
1.3 统计分析
广义遗传力H2=VG/(VG+VE),其中VG和VE分别为基因型变异和环境变异[27]。使用IciMapping软件利用群体基因型数据[23]构建遗传图谱,结合代谢组数据进行mQTL定位并计算每个位点的加性效应值。取LOD值大于2.5的位点为显著mQTL位点。热图使用R软件包绘制,网络图使用Cytoscape软件获得。
2 结果与分析
2.1 群体代谢谱检测结果
在群体样品中共检测到478种已知代谢物,其中154种物质通过与标准品比对鉴定得到。所检测到的物质主要包含氨基酸及其衍生物(Ami,50种)、黄酮类物质(Fla,72种)、脂质(Lip,44种)、核苷酸及其衍生物(Nuc,32种)、有机酸(Org,12种)、酚胺(Phe,29种)、植物激素及其衍生物(Phy,73种)、多酚(Pol,43种)、糖类(Sug,16种)、维生素及其衍生物(Vit,24种)以及其他代谢物(Oth,83种),见图1A。这些代谢物中广义遗传力介于0.4~0.9范围内的代谢物数量占比88.28%(图1B)。同时,有349种代谢物含量的变异系数为0.2~0.9,且有114种代谢物含量的变异系数大于0.9(图1C)。对这些代谢物含量分布规律分析显示,结构或者类别相似的部分代谢物被聚类在一起(图2A),例如包含吲哚环结构的一些代谢物在群体材料中具有类似的分布规律(图2B)。
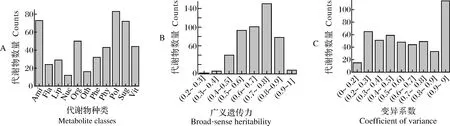
图1 代谢谱测定结果中代谢物种类 (A)、广义遗传力 (B) 以及变异系数 (C) 分布信息Fig.1 Distribution of metabolite classes (A),broad-sense heritability (B) and coefficient of variance (C)for each metabolite detected in the current study
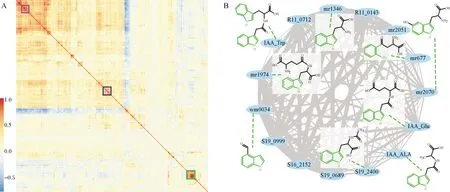
A:所有代谢物在小麦籽粒中分布规律的相关性热图,其中紫色、蓝色和绿色矩形方框标识所对应的代谢物具有相似分布规律,绿色方框内代谢物详情见图B; B:图A中绿色矩形方框对应的代谢物信息,这些代谢物之间分布规律的相关系数用灰色连线表示,连线越粗对应相关系数越高。这些高相关性代谢物中包含9种具有吲哚环结构(该结构骨架用绿色标识)的代谢物。A:Heatmap of correlation values regarding the similarities of metabolites distributed in wheat samples,within which the metabolites included in the colored (i.e.,purple,blue and green) rectangles have similar distribution patterns. The more detailed information of metabolites involved in the green rectangle are shown in Fig. 1B; B:Detailed information of metabolites included in the green rectangle of Fig. 1A. The Pearson correlation values are indicated by the grey lines,where higher correlation values correspond to thicker lines. The indole skeletons were commonly found in nine of the metabolites,and the skeletons are indicated by green.
2.2 代谢物数量性状位点(mQTL)定位与分析
利用IciMapping软件对RIL群体的SNP基因型所构建的遗传连锁图谱[24]和代谢物测定结果进行QTL分析,对236个代谢物共定位到385个mQTL位点(LOD > 2.5)。这些位点分布于小麦21条染色体上,其中2B和6B染色体定位到的mQTL位点最多,均为36个(图3A)。分析不同类别代谢物的定位结果数量,发现植物激素类物质(Phy)定位到的QTL位点数量更多,共有70个mQTL位点(图3B)。另外,不同类别的代谢物的平均定位位点数差异不大,而黄酮类(Fla)和酚胺类(Phe)代谢物最低,平均近2个代谢物才能定位到1个位点(图3B)。和其余类别代谢物相比,本研究中所检测到的大量黄酮类(Fla)和酚胺类(Phe)代谢物均没有定位到有效的QTL位点。除了2A和2D染色体的mQTL数量相等(图3A),mQTL数目在小麦3个亚基因组的分布整体呈现D基因组上mQTL数量更少的染色体偏好性。对于维生素类代谢物,在B基因组和D基因组所定位mQTL数量相等,而核苷酸类物质在D基因组上的位点数甚至超过B基因组(图3C)。此外,各类代谢物均呈现D基因组定位结果更少的染色体偏好性(图3C)。具体到每条染色体,有5类代谢物(氨基酸、核苷酸、有机酸、酚胺和糖类)在单条染色体mQTL数占该类物质所有位点数的20%以上(图3D),其中酚胺类物质接近50%的mQTL位点均位于5B染色体(图3D)。对定位到的所有mQTL位点进行汇总,发现共有5个位点为mQTL热点区域,这些位点均有不少于5个代谢物的定位结果(图4)。在这些热点区域中,共定位代谢物最多的位点为6B染色体短臂末端的0.0~0.5 cM区段(标记为RAC875_c1305_120和RAC875_c44002_81),共有22个代谢物共定位于该区段范围内。另外,位于5A染色体的热点2(BS00075308_51和Td_c94007_225)被5种核苷酸类物质共定位(图4);热点1(wExc15046_23216392和IAAV4072)被16种代谢物共定位,其中1个氨基酸类物质(S19_2400,N-Acetyl-tryptophan)的LOD值为20.9(图4A)。同时,该定位热点共包含5个含有吲哚环结构的色氨酸类似物(S19_2400、 mr677、mr1346、mr1974和mr2070,图4B),这些物质在该小麦群体中具有相似的含量分布规律(图1)。最后分析这5个定位热点区域对应位点的加性效应来源,发现其中3个热点(热点1、3、4)来源于亲本Naxos, 2个热点(热点2、5)则来源于Soru#1。

A:mQTL在不同染色体上的数量分布; B:不同类别代谢物所定位mQTL数量信息; C:不同类别代谢物在不同亚基因组上的mQTL分布; D;不同类别代谢物在21条小麦染色体上的mQTL分布。A:Distribution of mQTL on different chromosomes; B:mQTL numbers from each metabolite category; C:Distribution of mQTL on the three sub-genomes; D:Distribution of mQTL on the 21 wheat chromosomes.

5个mQTL热点分布在4个连锁群上(A),每个热点均定位到不同类别的代谢物类型(B)。热点1中所定位到的氨基酸类物质为5种色氨酸类似物(B),其中1种物质(S19_2400)定位的LOD值为20.9(A)。The five mQTL hotspots are distributed on four linkage groups (A),numerous metabolites classified into different categories were co-mapped on each of the hotspots (B). The amino acid derivatives mapped in hotspot 1 were five tryptophan-decorative metabolites (B),in which one (S19_2400) was mapped with LOD value of 20.9 (A).
3 讨 论
本研究通过对Soru#1与Naxos构建的高代RIL群体进行代谢组测定和定位,成功获得了大量的mQTL位点,这些信息将为未来解析该群体中代谢组差异的分子机制提供参考。同时,由于2个亲本在多种病虫害抗性上存在显著差异[23-24],相关定位结果还能与抗性定位结果进行联合分析,以便挖掘小麦抗病与代谢物之间可能存在的关联。本研究通过分析代谢谱,发现处于相同通路、结构类似的代谢物可能具有相似的含量分布规律(图2),该结果与小麦中已有报道[21]类似,相关结果有助于代谢物候选基因鉴定和通路解析。通过统计mQTL结果,发现代谢物定位结果的染色体偏好性(图3),这种偏好性有可能是由于小麦D基因组遗传多样性相对于A或者B基因组更低[28]所导致的。所获得的定位结果中还包含5个可能控制多种代谢物含量的热点区域(图4),在这些热点区域内可能存在同时控制多个代谢物含量的候选基因[25,29]。例如,位于5A染色体的热点2仅被6种核苷酸类物质共同定位(图4),该区域内可能包含影响多种核苷酸代谢物的候选基因;与此同时,5A染色体的热点1不仅对于代谢物S19_2400有较大LOD值,还被另外4种包含吲哚环结构的代谢物共同定位。由于这些代谢物结构类似(图1B)且处于色氨酸代谢途径,该染色体区段可能存在候选基因,主要通过控制代谢物S19_2400的合成或者分解,影响多种与之结构相关、处于相同代谢通路的色氨酸类物质的相对含量。通过分析所获得5个mQTL热点的加性效应来源,发现2个亲本对其中不同热点均有贡献,表明合理利用不同亲本组合有助于更好地进行小麦代谢组学研究,以及针对这些热点区域的进一步研究有助于候选基因鉴定和解析该群体代谢组差异。
