CARD11基因非经典区域突变所致免疫缺陷2例的临床和免疫特征
2021-06-07李天慈王文婕应文静刘璐瑶王晓川孙金峤
李天慈 王 莹 王文婕 应文静 刘璐瑶 胡 榆 王晓川 吕 伟 孙金峤,3
1 病例资料
1.1 病史报告
例1 男,6岁,因“反复呼吸道感染5年”于复旦大学附属儿科医院(我院)初次就诊。1岁后,无明显诱因出现反复呼吸道感染,表现为支气管炎、化脓性扁桃体炎,伴发热,热峰39℃以上,通常持续3~5 d,每月1次,偶伴关节痛、腹痛;3岁起,呼吸道感染频率增加,1~2周1次,予细菌溶解产物、胸腺肽等治疗无好转。体格检查:双侧颈部可触及数枚淋巴结,直径0.5~0.7 cm,质中,活动度可,无触痛,边界清楚,其他查体和体征未见异常。实验室检查提示,IL-6最高达79.8(参考值<7)pg·mL-1,RBC、Hb和PLT均正常,自身抗体阴性,EB病毒VCA IgG滴度为177(参考值为<20)RU·mL-1,EB病毒 DNA水平低于检测值下限。胸部CT未见异常,腹部B超腹腔内未探及肿大淋巴结。
患儿系G4P2,无反复发热病史。患儿母亲第1和2胎均不明原因早期流产;第3胎系患儿姐姐,无反复感染、发热病史,未行基因检查。随访患儿至8岁时诊断为变应性鼻炎,行颞骨CT,提示部分鼻黏膜增厚。
例2 男,7岁,10月龄起无明显诱因出现反复呼吸道感染伴发热,体温最高达40℃,通常持续5~7 d,伴咳嗽,无气喘,偶伴呕吐,无腹痛、腹泻,抗感染治疗后可好转;3~4岁感染最为严重,1~2个月1次呼吸道感染伴发热,胸腺肽治疗后发病频率较前下降;6岁起出现特应性皮炎,头皮可见散在硬币大小斑秃样皮损,伴瘙痒,夜间加重。查过敏原提示对鸡蛋、尘螨过敏。体格检查未见异常。患儿父亲自幼反复呼吸道感染伴发热病史,学生时期最为严重,严重时呼吸道感染每月1次,成年后呼吸道感染每年3~4次,每次持续5~7 d,抗生素治疗有效。母亲体健。
1.2 免疫相关检查
1.2.1 免疫球蛋白 外周血IgA、IgG及IgM浓度测定采用免疫透射比浊法,外周血IgE浓度采用UniCAP诊断系统检测。表1显示,例2 IgE水平显著增高。
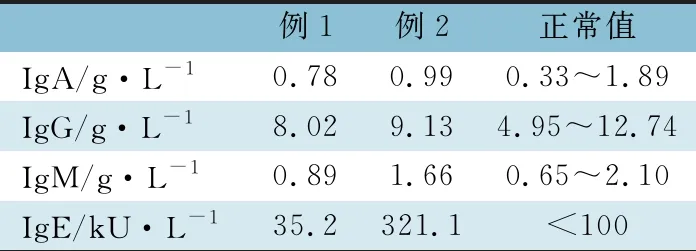
表1 2例患儿的免疫球蛋白水平
1.2.2 外周血免疫细胞分型 2例血常规显示外周血淋巴细胞分别为60%和72%,绝对计数分别为3 100和5 200个/μL,提示存在淋巴细胞过度增殖现象。
1.2.3 外周血淋巴细胞分型及精细分型 ①淋巴细胞分型:免疫荧光法检测红细胞裂解后的外周血淋巴细胞的比例及绝对数(BD公司的Multitest IMK试剂盒)。表2显示,例1 CD3+T细胞比例升高,CD8+T细胞数量、比例轻度升高;例2 T细胞计数及比例显著降低,B细胞计数及比例显著升高。 ②淋巴细胞精细分型:取患儿肝素抗凝外周血50 μL,加入流式抗体后室温避光孵育30 min,加入红细胞裂解液,水浴离心,弃上清,加入适量磷酸盐缓冲液后上机(BD FACSCanto Ⅱ);图1显示,2例过渡B细胞比例均显著升高。例2初始B细胞比例显著升高,记忆B细胞比例显著下降;CD4+初始T细胞比例上升, CD4+中枢记忆T细胞及CD4+效应记忆T细胞比例下降;CD8+初始T细胞比例升高,CD8+中枢记忆T细胞和CD8+效应记忆T细胞比例下降。
1.3 基因检测结果 对患儿及其父母行全外显子测序(WES)。取患儿及其父母外周血,提取基因组DNA,经超声打断、末端修复、接头连接和杂交捕获构建文库,采用Illumina公司HiSeq测序平台进行序列检测,以人类基因组hg19(GRCh37)为参考序列进行比对。2例患儿7号染色体CARD11基因(NM_032415)均存在杂合突变,例1突变位点:exon19的c.2542 C>T(p.R848C),突变来自母亲;例2突变位点:exon16的c.2036 A>T(p.Q679L),突变来自父亲,既往未见报道。
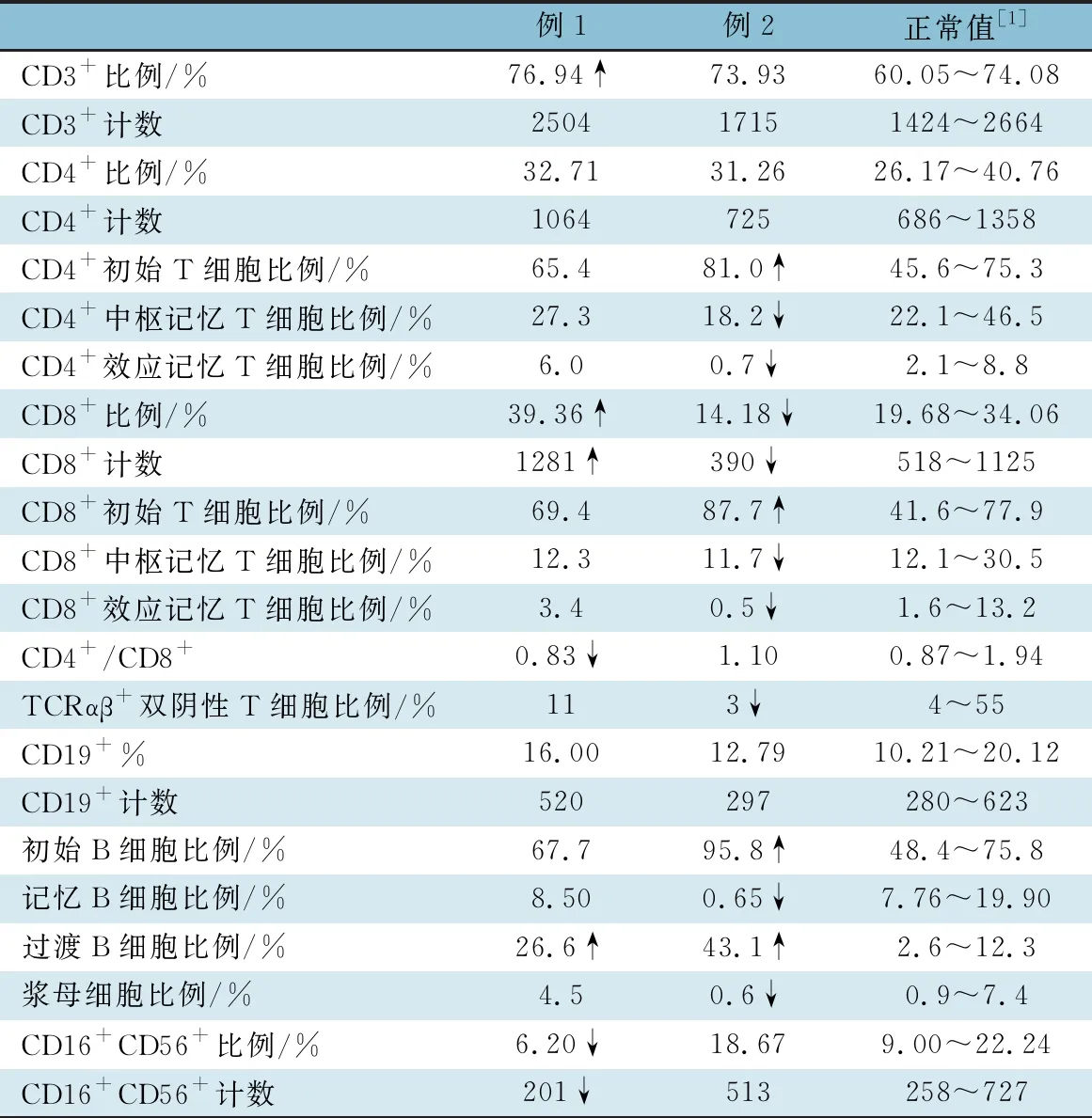
表2 2例患儿外周血淋巴细胞比例及绝对数量
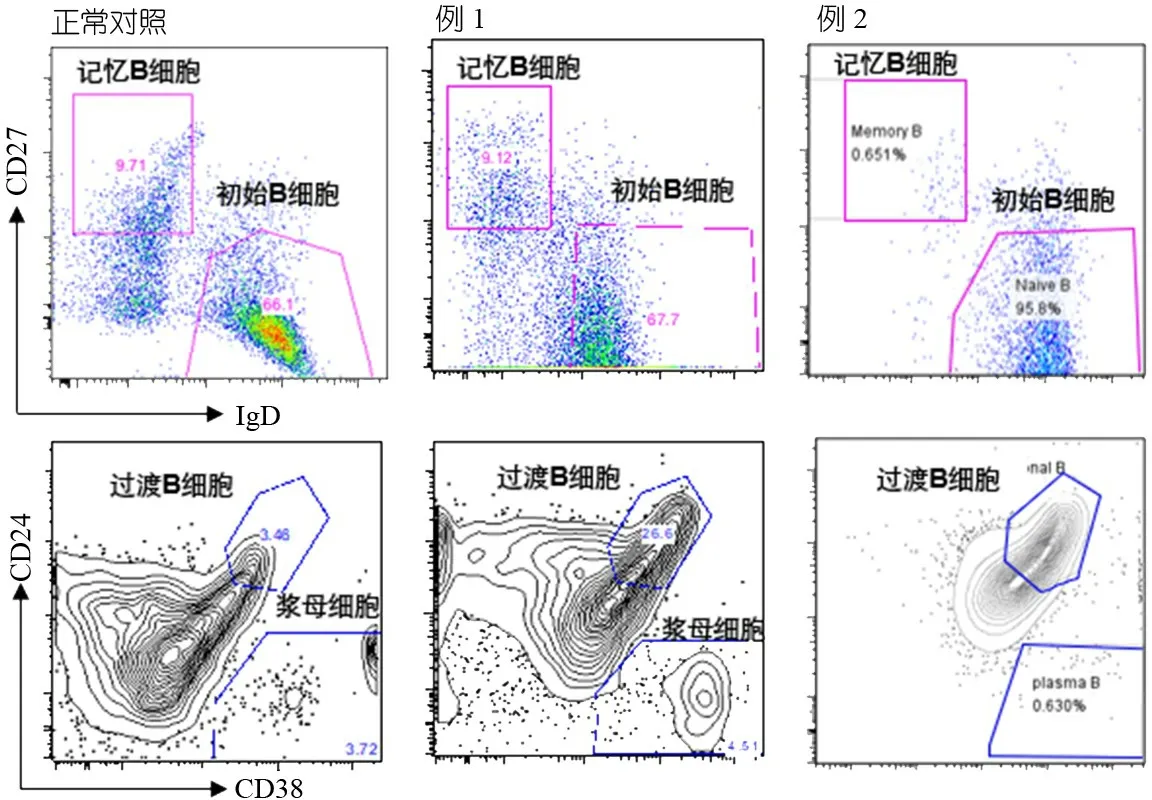
图1 2例患儿外周血B淋巴细胞精细分型结果
取患儿及父母外周血基因组DNA,设计引物进行PCR扩增(2 ×Multiplex PCR Mix,上海近岸科技有限公司)。突变验证引物序列如下。例1正向:CCATCCCTACGCAGAG-TCG,反向:GGTTTGAAGCTGGCGAGATC;例2正向:ACATTCTGTTTCCTGCTGCA,反向:GACACTACAGTTGCAGCCAC。Sanger验证结果见图2。患儿突变分别导致CARD11蛋白第848位点的精氨酸突变为半胱氨酸(例1)以及第679位点的谷氨酰胺突变为亮氨酸(例2)。2个突变PhyloP得分分别为0.758和4.144(-14~6,正值代表保守),PhastCons得分均为1分(0~1,越接近1则保守度越高),说明在不同物种间这2个突变位点及其附近序列高度保守。经mutation taster网站预测,2个突变均为致病突变,氨基酸改变得分分别为180和113。2个突变的PROVEAN得分[1, 2]分别为-1.954和-3.134,预测结果亦为致病突变。ExAC及1000G数据库中未发现c.2036 A>T。
CARD11基因突变导致的疾病以功能获得性点突变导致B细胞增殖伴NF-κB激活、T细胞无能疾病(BENTA)及弥漫大B细胞淋巴瘤(DLBCL)最具代表性。这类功能获得性CARD11突变通常位于CARD11基因的CARD结构域、CC结构域及其之间的部分。图3显示,例1携带的突变位于SH3结构域及GUK结构域之间,例2携带的突变位于PDZ结构域,与经典的CARD11突变相比在基因中的位置相对靠后。
1.4 体外NF-κB通路激活实验结果 体外使用Gibco DMEM培养基,加入10%胎牛血清、1%谷氨酰胺、1%青霉素,在37℃、5% CO2培养箱中培养293T细胞。使用聚乙烯亚胺(PEI)为媒介,转染293T细胞。12孔板培养细胞,细胞密度70%左右,每孔转NF-κB GFP报告质粒及目的质粒各0.5 μg,6 h后换液。图4显示,24 h后检测GFP表达情况[NF-κB通路激活时NF-κB GFP 报告质粒可表达绿色荧光蛋白(GFP),表达情况与通路激活程度成正比],与野生型相比,在293T细胞系中过表达患儿涉及的2种突变蛋白,表达GFP的细胞比例及GFP表达的平均荧光强度所反映的NF-κB通路的激活情况均减弱,提示患儿所携带的2种突变均导致功能缺失。

图2 2例CARD11基因突变患儿一代测序验证结果及家系图

图3 文献报道[4-8]的致BENTA及DLBCL的CARD11突变
及本文患儿突变在基因中的位置
注 黑色为致BENTA的CARD11突变,灰色为致DLBCL的CARD11突变,均位于基因靠近N端的CARD及CC结构域中。红色为本文患儿携带的突变位点

图4 在293T细胞中过表达野生型(WT)及突变的CARD11基因后NF-κB通路的激活情况
2 讨论
Caspase 募集结构域家族成员11(CARD11)基因编码的同名蛋白CARD11是一种含半胱天冬酶募集结构域的支架蛋白,属于膜相关鸟苷酸激酶家族,其表达具有淋巴细胞特异性[3]。在适应性免疫中,尤其是T细胞受体(TCR)介导的NF-κB途径激活中,CARD11蛋白发挥着重要作用[3]。在TCR和B细胞受体(BCR)接受刺激后,CARD11蛋白发生构象改变,与黏膜相关淋巴组织淋巴瘤易位蛋白1(MALT1)以及B细胞淋巴瘤/白血病10(BCL10)组成CBM复合物,充当细胞间联系的桥梁,传递细胞表面的受体信号,激活下游的NF-κB通路,在适应性免疫中发挥重要作用[3]。
CARD11缺陷的临床表现复杂多样,杂合的功能获得性种系突变可以导致一种名为BENTA的原发免疫缺陷病。1971年Darte等[4]描述了第1例CARD11突变导致BENTA的患儿,表现为反复呼吸道感染、脾肿大以及外周血B细胞中过渡性B细胞比例显著升高。Arjunaraja等[5]发现导致BENTA的CARD11突变还可影响B淋巴细胞的分化,表现为刺激后短期浆细胞及长期浆细胞比例的显著降低。类似的功能获得性点突变若发生在体细胞中,则可导致弥漫性大B细胞淋巴瘤[6, 7]。纯合的功能缺失性突变可以导致免疫缺陷病11A,表现为自幼出现的反复感染,以肺炎最为常见;患儿T、B细胞数正常但免疫球蛋白水平降低,外周血淋巴细胞分型以幼稚或过渡性T、B淋巴细胞增高,调节性淋巴细胞[8]及记忆细胞减低为主要特征[9]。另外,杂合的功能缺失点突变亦可导致伴变应性皮炎的免疫缺陷病11B,呈常染色体显性遗传。Ma等[10]发现此病患者表现为轻重不一的特应性皮炎,同时伴不同程度的感染,如一过性或反复发作的肺炎、软疣、脓肿、菌血症等,部分患者可出现外周血IgE水平升高及嗜酸性粒细胞计数增多,还可出现一过性的低丙种球蛋白血症、外周血IgM水平降低。
本文2例患儿均有反复感染临床表现和外周血过渡B细胞显著增高的CARD11突变导致免疫缺陷的特征性免疫表型,突变致病性预测提示,携带的突变均致病,提示患儿所患疾病应与CARD11突变有关。例1携带的突变,Dorjbal等[11]认为其为致病突变,精细分型的检查进一步印证了此突变的致病性。例2基因型及临床表型均符合CARD11突变致病表现,且疾病表现为常染色体显性遗传,其父亲具有相似临床特征,进一步证实了突变与疾病之间的关联性。目前已报道的功能获得性CARD11突变,主要位于基因N端的CARD、CC结构域或2个结构域之间的部分(有文献称这部分为LATCH结构域)[6, 12]。这些突变使NF-κB通路持续激活,从而导致了疾病的发生[13]。而导致免疫缺陷病11A和伴变应性皮炎的免疫缺陷病11B的功能缺失性点突变,则不会出现NF-κB通路的自发激活[9]。本文例1突变位于SH3结构域与GUK结构域之间,例2突变位于PDZ结构域,均距CC结构域较远。仅有少量涉及GUK结构域附近的功能缺失性点突变的文献报道,关于PDZ结构域的致病突变尚未见报道。结合功能实验,患儿所携带的点突变不会导致NF-κB通路的自发过度激活的结果,2例突变应为功能缺失性点突变。与经典的BENTA相比,本文2例B细胞克隆增殖程度较轻,脾脏均无肿大,无B细胞淋巴瘤,应归类为伴变应性皮炎的免疫缺陷病11B。
本文涉及的实验及文献中报道的研究结果,均提示这些功能缺失性突变的致病机制可能不依赖NF-κB通路,且致病突变在基因中的相对位置并不局限在经典的导致BENTA或DLBCL的结构域,提示C端的结构域对于CARD11基因功能的发挥亦有重要作用,除NF-κB通路外,CARD11基因还可能通过其他通路影响免疫功能。
值得注意的是,例1母亲携带与患儿相同的致病突变,但无显著的反复感染伴发热病史,Buchbinder等[12]报告一类似家系,即相同基因型,男孩患病但母亲无显著临床表现,提示CARD11突变导致的免疫缺陷可能存在不完全显性。例2与其父亲有相同基因型及临床表型,结合与CARD11突变密切相关的弥漫性大B细胞淋巴瘤存在一定的性别倾向,尤其是在年轻患者中男性多于女性,治疗结果也受性别影响[14, 15],提示CARD11突变致病的临床表现与严重程度可能与性别存在相关性。
